
Abstract
Inflammatory cells and mediators form a major part of the tumor microenvironment and play important roles in the regulation of cancer initiation, tumor cell proliferation, and metastasis. MicroRNAs (miRNAs) play important roles in several physiological and pathological processes, including the regulation of the inflammatory microenvironment in cancer. Transforming growth factor-β (TGF-β) is an inflammation-related cytokine that functions in both tumor suppression and promotion; however, its underlying molecular mechanisms remain unclear. Recent evidence indicates an association between miRNAs and TGF-β signaling, providing new insight into the nature of the inflammatory microenvironment in cancer. The present review is an overview of the interaction between miRNAs and inflammatory cytokines, with emphasis on the cross talk between TGF-β signaling and miRNAs and their influence on cancer cell behavior. The emerging roles of miRNAs in cancer-related inflammation and the potential to target miRNA signaling pathways for cancer therapy are also discussed.
Similar content being viewed by others
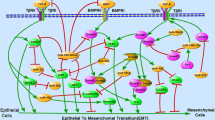


Introduction of miRNA and the tumor inflammatory microenvironment
MicroRNAs (miRNAs) are endogenous single-stranded non-coding RNAs of 18–25 nucleotides [1] that function in the post-transcriptional regulation of gene expression through translational repression or cleavage of messenger RNA (mRNA) targets in a specific base pairing manner [2]. One miRNA has the potential to affect the expression of several proteins, and one protein can be regulated by several miRNAs. The modulation of oncogenic and anti-oncogenic miRNAs could, in principle, affect the progression of cancer [3–5]. miRNA expression profiling of human tumors has identified signatures associated with diagnosis, staging, progression, prognosis, and response to treatment [6]. Emerging evidence suggests a direct link between miRNAs and cancer [7], inflammation [8, 9], and autoimmune diseases [10–12]. miRNAs regulate immune responses by modulating the expression of immune-related genes.
The connection between cancer and inflammation was first identified in the nineteenth century and is currently recognized as one of the six hallmark features of cancer development and progression [13, 14]. The field of cancer-related inflammation is driven by the hypothesis that extrinsic pathways associated with conditions that cause or promote cancer and intrinsic inflammatory pathways activated by genetic events lead to the production of inflammatory cytokines [13, 15]. Inflammatory cells and inflammatory mediators form a major part of the tumor microenvironment, which has been highlighted as an important factor in cancer progression (Fig. 1). For example, CD4(+)CD25(high)Foxp3(+) regulatory T cells (Tregs) enriched in the tumor-associated microenvironment play an important role in cancer immune evasion [16] and are considered potential therapeutic targets in human diseases [17]. However, the complex nature of the inflammatory microenvironment remained unclear until recently, when several miRNAs, such as miR-126, miR-132, miR-146, miR-155, and miR-221, emerged as important transcriptional regulators of inflammatory mediators [8]. Research in the miRNA field opened new horizons in our understanding of the role of the inflammatory microenvironment in cancer. In the present review, we focus on the interaction between miRNAs and inflammatory cytokines in the tumor microenvironment, with particular emphasis on the transforming growth factor-β (TGF-β) signaling pathway. In addition, we present a novel viewpoint based on the modulation of the tumor microenvironment as miRNA-based cancer therapy.

Overview of the interaction of cytokines in the inflammatory tumor microenvironment. Tumor-associated inflammation develops simultaneously with tumor development. The tumor microenvironment is a complex scaffold of various cells and extracellular matrix. Different cells contribute by producing cytokines and respond to stimuli secreted by other cells. This creates a favorable microenvironment for tumor growth and progression. The inflammatory response plays a central role in angiogenesis, tumor proliferation, local immunosuppression, and metastasis
Control of miRNA biogenesis by inflammatory cytokines
Originally, research aimed at investigating miRNA expression profiles revealed differences in the expression of specific miRNAs in cancers. Recently, research has started to focus on the regulation and function of miRNAs. Several studies have investigated the specific influence of key inflammatory cytokines on miRNA expression. Overexpression of inflammatory cytokines such as tumor necrosis factor (TNF)-α, interleukin (IL)-6, IL-1, IL-8, IL-10, IL-12, and TGF-β can either promote or inhibit cancer development [18, 19]. Several miRNAs such as miR-155 and miR-21 have been implicated in cancer development and inflammation [20], and they are controlled by inflammatory mediators, the most prominent being Toll-like receptors (TLRs), TNF, TGF-β, and other cytokines that link the functions of miRNAs with inflammatory events [11, 20–24]. Table 1 describes the miRNAs implicated in both cancer and inflammation and their functions.
Aberrant miRNA expression leads to developmental abnormalities and diseases; however, the processes regulating miRNA biogenesis are largely unknown. miRNAs are transcribed as long and capped polyadenylated pri-miRNAs. The pri-miRNA is cropped into a hairpin-shaped pre-miRNA by the Drosha complex. Next, the pre-miRNA is translocated to the nucleus by exportin-5 and further processed by the Dicer complex. The resulting miRNA is dissociated and incorporated into the RNA-induced silencing complex (RISC), where it functions in gene silencing by promoting the degradation of target mRNAs or by translational inhibition. The identification of mechanisms of miRNA biogenesis regulation revealed that various factors or growth factor signaling pathways control every step of the miRNA biogenesis pathway [52].
miR-155, which was the first miRNA shown to play an oncogenic role [53], is overexpressed in a variety of tumors such as leukemia/lymphoma, breast, colon, lung, pancreatic, and gastric tumors [25, 26]. Enhanced expression of miR-155 is often associated with increased cytokine expression, a prominent feature of inflammatory processes [27, 54, 55]. For example, lipopolysaccharide (LPS)/TNF-α stimulation results in the upregulation of miR-155 and downregulation of miR-125b [27]. miR-21 is upregulated in almost all carcinomas and hematological malignancies [6] and is induced in macrophages and blood mononuclear cells upon lipopolysaccharide (LPS) challenge [34] and in mammary epithelial cells by inflammatory signals [35]. miR-210 links inflammatory signals with the hypoxic microenvironment, as it is induced in response to low oxygen and inhibited by the cytokines IL-6 and TNF [42]. On the other hand, inflammatory cytokines can be modulated by miRNAs. Several miRNAs, such as miR-126, miR-132, miR-146, miR-155, and miR-221, are important transcriptional regulators of certain inflammation-related mediators [8].
Upregulated miRNAs by the TGF-β/Smad signaling pathway
TGF-β function is dependent on tissue type and the epigenetic background of cells [56]. One prominent feature of TGF-β biology is its dual role: It functions as a tumor suppressor in the early stages of tumorigenesis, whereas it promotes tumor cell metastasis in advanced stages [56, 57]. The interaction between TGF-β signaling and miRNAs has been investigated extensively, and studies suggest that the TGF-β pathway can either inhibit or enhance miRNA maturation [58–61]. Figure 2 shows a brief outline of the miRNAs associated with the TGF-β/Smad signaling pathway in cancer.

The TGF-β signaling pathway and related miRNAs. Ligand binding to the TGF-β receptor (TβR)-mediated heterotetramer formation. This signal is relayed through the nucleus via restricted Smad (R-Smad). Activated R-Smads complex with common Smads (Co-Smad, such as Smad-4) translocate to the nucleus and alter miRNA transcription. In addition, TGF-β signaling alteration of miRNA transcription may depend on non-Smad molecules, such as ERKs, JNK/p38, RhoA, and PI3K/Akt. The mechanisms of TGF-β signaling-mediated miRNA expression are either transcriptional or post-transcriptional
Davis et al. [62] was the first to describe mechanisms of miRNA expression modulation and showed that TGF-β treatment resulted in the upregulation of pre-miRNAs and mature miRNAs, but not that of pri-miRNAs. Smad proteins have been shown to control the transcription of miRNA-coding genes by binding to miRNA promoter genes [59]. Smads control miRNA biogenesis by two different mechanisms that involve complex Smad2–3 binding to Smad4 or not. The Smad2–3 complex is translocated to the nucleus, where it is recruited by the Drosha/DGCR8 microprocessor complex and promotes miRNA maturation [23, 58, 59, 63, 64]. However, the mechanism underlying the translocation of the Smad2–3 complex to the nucleus remains undetermined. The most prominent miRNAs upregulated by TGF-β signaling are miR-21, miR-181, miR-494, and miR-10.
miR-21
A meta-analysis revealed that high miR-21 levels are related to poor overall survival, suggesting that miR-21 is a prognostic factor for poor survival in cancer patients [65]. TGF-β-mediated induction of miR-21 was firstly found to be independent of Smad4, although it is dependent on the R-Smads [62]. TGF-β-induced miR-21 levels are markedly higher in transformed cells, and miR-21 contributes to chemoresistance in breast cancer cells by targeting the MutS homolog 2 [66]. In bladder tumors, in accordance to the observed similarity between TGF-β variants and miR-21 gene expression alterations, treatment of the bladder cancer cell line 5637 with recombinant TGF-β induced a significant upregulation of miR-21 [67] and confirmed the correlation between TGF-β and miR-21 expression in cancer. Furthermore, miR-21 is regarded as a potential therapeutic target [68]. Wang et al. [69] investigated the effect of ursolic acid (UA) on human gliomas and found that UA inhibits cell growth by inducing apoptosis in U251 cells via a UA-triggered TGF-β1/miR-21/PDCD4 pathway.
miR-181
Wang et al. [70] demonstrated that TGF-β-mediated upregulation of hepatic miR-181b promotes hepatocarcinogenesis by targeting the tissue inhibitor of metalloprotease 3 (TIMP3) [70]. In this article, the authors showed that the upregulation of hepatic TGF-β and its downstream Smad mediators in liver nuclear extracts correlated with elevated miR-181b/d in mice fed a choline-deficient and amino acid-defined (CDAA) diet. The levels of miR-181b increased upon exposure of hepatic cells to TGF-β and were inhibited by siRNA-mediated depletion of Smad4, demonstrating the role of TGF-β signaling in miR-181b biogenesis. Moreover, miR-181a had a direct effect on inducing hepatocyte epithelial-mesenchymal transition (EMT) and was able to replace TGF-β-induced effects in vitro [71]. In the field of breast cancer research, Wang et al. [72] observed that exposure to TGF-β increased the population of breast cancer cells that can form mammospheres in suspension, and this was mediated by miR-181, which was upregulated by TGF-β at the post-transcriptional level. Neel et al. [73] identified miR-181 as a potent regulator of TGF-β signaling in human breast cancer and found that miR-181 is the Smad2/3-dependent downstream target of TGF-β signaling. Furthermore, their data demonstrates that miR-181 is required for TGF-β-mediated cell migration and invasion, as silencing miR-181 expression significantly antagonizes the pro-invasive effects of this growth factor.
miR-494
Myeloid-derived suppressor cells (MDSCs) potently suppress the anti-tumor immune responses that favor tumor angiogenesis and metastasis; however, the molecular networks regulating the accumulation of tumor-expanded MDSCs are largely unknown. Liu et al. [74] revealed that TGF-β-upregulated miR-494 is required for the accumulation and activity of MDSCs via targeting of phosphatase and tensin homolog (PTEN). In pancreatic ductal adenocarcinomas (PDACs), dysregulation of β-catenin and the transcriptional activator FOXM1 mediates oncogenesis, although the underlying mechanism remained unclear until Li et al.[75] showed that loss of Smad4 in PDAC cells leads to reduced levels of miR-494, increased levels of FOXM1, and nuclear localization of β-catenin. Therefore, miR-494 might be developed as a prognostic marker for patients with PDAC or as a therapeutic target.
miR-10b
Ma et al. [76] firstly reported miR-10b upregulated in breast cancer and showed that the level of miR-10b expression in primary breast carcinomas correlates with clinical progression. Then, the results of follow-up studies supported these results [77, 78]. Recently, miR-10b is identified as a target gene of TGF-β1, which upregulated miR-10b, promotes EMT, and increases the metastatic potential of breast cancer cells [79]. Ouyang et al. [80] demonstrated that overexpression of miR-10b accelerated pancreatic cancer cell (PCC) proliferation and tumor growth in an orthotopic model. In this article, the authors showed that miR-10b and TGF-β function together to markedly increase cell invasion, and this effect is blocked by the combination of erlotinib and SB505124, a type I TGF-β receptor inhibitor. miR-10b also enhances the stimulatory effects of TGF-β on cell migration and EMT. Therefore, therapeutic targeting of miR-10b in malignancy may block growth-promoting and antagonize the metastatic process at various levels.
Other microRNAs: miR-27a/miR-183/miR-182/miR-155/miR-451
TGF-β-associated miR-27a inhibits dendritic cell-mediated differentiation of Th1 and Th17 cells by modulating TAB3, p38 mitogen-activated protein kinase (MAPK), MAP2K4, and MAP2K7 [81]. TGF-β-inducible miR-183 silences tumor-associated natural killer cells [82]. TGF-β-induced miR-182 induces an aggressive phenotype by targeting CYLD in glioma subsets [83]. TGF-β upregulates miR-155 through Smad4 and contributes to epithelial cell plasticity by targeting RhoA in breast cancer [84]. Moreover, Smad upregulates miR-451 and drives glioblastoma stem cell to no-stem cell transformation, leading to reduced glioblastoma tumorigenicity [85].
Downregulated miRNAs by the TGF-β/Smad signaling pathway
With respect to cancer, miRNAs are often located in fragile genomic sites and are therefore typically downregulated in tumors [6]; inhibition of miRNA biogenesis tends to enhance tumorigenesis [86], raising the possibility that miRNA re-expression potentially represents an effective therapy for cancer [87]. The examples below are representative miRNAs downregulated by TGF-β signaling.
miR-200
Gregory et al. [88] firstly reported that all five members of the miR-200 family (miR-200a, miR-200b, miR-200c, miR-141, and miR-429) are markedly downregulated in cells that undergo EMT in response to TGF-β. These authors showed that these miRNAs cooperatively regulate the expression of the E-cadherin transcriptional repressors zinc finger E-box binding homeobox 1 (ZEB1; also known as deltaEF1) and SIP1 (also known as ZEB2), factors previously implicated in EMT and tumor metastasis. Truong et al. [89] later found that TGF-β-downregulated miR-200 elicits migratory behavior by increasing ZEB2 in triple-negative breast cancer. Recently, a TGF-β-miR200-Mig6 pathway was shown to induce resistance to EGFR inhibitors in lung and pancreatic cancers [90].
miR-34a
Altered miR-34 expression has been determined to be involved in the pathogenesis of many cancers [91]. The miR-34a is one member of miR-34 family, which of miR-34a, miR-34b, and miR-34c. miR-34a is encoded by its own transcript, whereas miR-34b and miR-34c share a common primary transcript. Ectopic re-expression of miR-34a in both primary and tumor-derived cell lines is correlated with cycle arrest, apoptosis, and cell growth inhibition [92]. Yang et al. [93] demonstrated that elevated TGF-β activity associated with the persistent presence of hepatitis B virus in liver tissues suppresses the expression of miR-34a, leading to enhanced production of the chemokine CCL22, which recruits regulatory T cells to facilitate immune escape. This indicates that restoring the tumor suppressor miR-34, as well as blocking TGF-β signaling, may provide a novel molecular therapy for the treatment of cancer.
miR-203
miR-203 has been identified as a skin-specific keratinocyte-derived miRNA that is located at chromosome 14q32-33 [94]. miR-203 expression is significantly downregulated in laryngeal squamous cell carcinoma and is correlated with poor differentiation, advanced clinical stage, and lymph node metastasis [95]. Xu et al. [96] revealed that miR-203 is downregulated in renal cancer and confirmed that FGF2 is a direct target of miR-203. Ding et al. [97] demonstrated that TGF-β represses the expression of miR-203 to promote EMT and tumor metastasis. In this paper, miR-203 was significantly downregulated in highly metastatic breast cancer cells, and restoration of miR-203 in highly metastatic breast cancer cells inhibited tumor cell invasion in vitro and lung metastatic colonization in vivo by repressing SNAI2.
miR-584
miR-584 was shown to decrease cell motility through the inhibition of Rho-associated coiled-coil-containing protein kinase 1 (ROCK-1) in renal cell carcinoma (RCC) cell lines, and the expression of miR-584 was inversely correlated with that of ROCK-1 in RCC tissues [98]. These results suggest that miR-584 functions as a new tumor suppressor miRNA in RCC by downregulating ROCK-1. Fils-Aime et al. [99] identified miR-584, a potential tumor suppressor, as a novel target of TGF-β and found that miR-584 expression is negatively regulated by this growth factor in a number of breast cancer cells. These authors found that inhibition of miR-584 expression by TGF-β is required for cell migration, as overexpression of ectopic miR-584 reversed TGF-β-induced cell migration. They further identified protein phosphatase and actin regulator 1 (PHACTR1) as a downstream target of miR-584 and found that TGF-β-mediated inhibition of miR-584 leads to increased expression of PHACTR1.
miR-450b-5p
TGF-β1 was shown to exert its function by suppressing miR-450b-5p, which significantly inhibited the growth of rhabdomyosarcoma (RMS) and promoted the expression of MyoD. Using a bioinformatics approach, we identified ecto-NOX disulfide-thiol exchanger 2 (ENOX2) and paired box 9 (PAX9) as targets of miR-450b-5p. These results suggest that disrupting the TGF-β1 suppression of miR-450b-5p or knockdown of ENOX2 and PAX9 are effective approaches in inducing RMS MyoD. Furthermore, we found that the Smad3 and Smad4 pathways, but not Smad2, are the principal mediators of TGF-β1 suppression of miR-450b-5p [100].
miRNAs regulate the TGF-β signaling pathway
Most members of the TGF-β pathway are known to be targeted by one or more miRNAs [23]. After the identification of TGF-β1, TGF-βR (I and II) and Smads were found to be dysregulated in most cancers, whereas miRNAs potentially targeting these molecules are downregulated. The impact of miRNA on canonical Smad signaling has been investigated extensively.
TGF-β1
Interactions between miRNAs and TGF-β1 signaling have been validated experimentally with suggesting that miRNAs influence the TGF-β1 signaling at multiple levels. A few such interactions have been demonstrated. For example, Martin et al. [101] identified multiple binding sites for miR-744 located in the proximal TGF-β1 3′-UTR. miR-744 transfection inhibited endogenous TGF-β1, which given the pleiotropic nature of cellular responses to TGF-β1 is potentially significant. Dogar et al. [102] showed that reduced levels of the oncomirs miR-18a and miR-24 accounted for the observed derepression of two TGF-β1 processing factors, thrombospondin-1 (THBS1) and furin, respectively, which suggested a novel mechanism by which latent TGF-β1, thrombospondin 1, and furin form a miRNA-mediated regulatory feedback loop. Ectopic expression of latent TGF-β1 reduces THBS1 protein expression and is associated with increased expression of let-7 and miR-18a in cells [103]. These data suggest an inverse correlation between THBS1 and latent TGF-β1 expression levels possibly involving miRNAs.
TGF-βR I and II
Downregulation of miR-30 or miR-200 upregulates TGF-βR I and Smad2 to direct the EMT and invasive potential of anaplastic thyroid carcinomas [104]. In breast cancer cells, miR-128a promotes letrozole resistance by targeting TGF-βR I [105]. To date, many miRNAs targeting TGF-βR II have been identified in different malignancies and shown to contribute to tumor progression. miR-590-5p promotes the proliferation and invasion of human hepatocellular carcinoma cells [106], and miR-106b induces the migration and invasion of colorectal cancer cells [107]. High expression of miR-370 in gastric carcinoma tissues is associated with increased nodal metastasis and advanced clinical stage compared with controls [108], and miR-211 promotes the tumorigenesis of head and neck carcinomas [109]. Moreover, miR-21 induces stemness by targeting TGF-βR II in colon cancer cells [110] and promotes tumor development by targeting of TGF-βR II and Smad2/3 in prostate cancer [111]. The miR-520/373 family functions as a tumor suppressor in estrogen receptor negative breast cancer by targeting NF-κB and TGF-βR II [112]. Downregulation of miR-655 increases ZEB1 and TGF-βR II levels to accelerate cancer progression [113]. Recently, the miR-17 family was shown to reverse cisplatin resistance and suppress metastasis by targeting TGF-βR II in NSCLC [114].
Smads
miR-148a attenuates the cancer stem cell-like properties of hepatocellular carcinoma cells by targeting Smad2 [115]. miR-99a and miR-99b enhance cell migration and adhesion of normal murine mammary gland cells by targeting Smad3 [116]. miR-92b functions as a potential oncogene by targeting Smad3 and promotes glioblastoma cell proliferation [117].
miR-146b-5p increases cell proliferation and cell cycle arrest by repressing Smad4 in thyroid cancer [118]. miR-199a acts as a negative regulator of TGF-β signaling by targeting Smad4 and plays an oncogenic role in gastric cancer [119]. miR-130a-mediated downregulation of Smad4 reduces the sensitivity to TGF-β1 stimulation in granulocytic precursors [120]. Moreover, miR-130a/301a/454 functions as oncogenes by targeting Smad4 to enhance cell proliferation and migration in human colorectal cancer [121]. As a tumor suppressor miRNA, miR-34a acts as a tumor suppressor by targeting Smad4 in proneural subtype glioblastoma [122]. Downregulated miR-146a increases Smad4 and affects cell proliferation in response to retinoid acid induction in an acute promyelocytic leukemia cell line [123].
miR-155 renders diffuse large B cell lymphoma (DLBCL) cells resistant to the growth inhibitory effects of TGF-β1 and BMP by targeting Smad5 [124]. Moreover, miR-155 controls RB phosphorylation in normal and malignant B lymphocytes via the non-canonical TGF-β1/Smad5 signaling module [125]. Kaposi’s sarcoma-associated herpesvirus-encoded miR-K12-11 facilitates cell proliferation through the suppression of Smad5 [126]. The miR-106b-25 cluster targets the inhibitory Smad7 protein, resulting in the overexpression of TGF-βR I, and activates TGF-β signaling to induce EMT and the tumor-promoting effect of Six1 treatment in human breast cancer cells [127]. Moreover, miR-216a/217 induces EMT by targeting PTEN and Smad7 to promote drug resistance and recurrence of liver cancer [128].
Other mechanisms
The miR-106b-25/miR-17-92 cluster interferes with cell cycle arrest and apoptosis to resist TGF-β tumor suppression [129]. Moreover, the miR-106b-25 cluster inhibits the TGF-β tumor suppressor pathway, interfering with the expression of CDKN1A and BCL2L11 (Bim) in gastric cancer [130]. The miR-17-92 miRNA cluster was shown to act both upstream and downstream of psmad2, resulting in the downregulation of multiple key effectors of the TGF-β signaling pathway and in the inhibition of TGF-β-responsive genes in neuroblastoma cells [131]. miR-183 inhibits TGF-β1-induced apoptosis by downregulating PDCD4 in human hepatocellular carcinoma cells [132]. Moreover, miR-204 and miR-379 target IL-11 and downregulate the expression of several genes involved in TGF-β signaling, including PTGS2 in bone metastatic breast cancer cells [133]. miR-127-3p inhibits cancer cell proliferation and activates TGF-β signaling by targeting SKI in glioblastoma [134].
miRNAs and non-Smad pathways in malignancy
In addition to the canonical Smad pathway, the effects of TGF-β are largely dependent on the interactions between numerous non-canonical signaling factors such as ERK, p38, RhoA, and phosphoinositide 3 kinase (PI3K) [135, 136]. The effect of the canonical signaling pathway on miRNA expression regulation has been investigated and was reviewed by Blahna et al. [58]. However, miRNA alterations of non-canonical signaling in malignancies have been characterized to a lesser extent. Since differences in miRNA expression levels will impact non-canonical signaling, these different miRNAs could exert anti-tumor effects by altering the TGF-β signaling pathway, highlighting miRNAs targeting non-canonical signaling as potential therapeutic targets. For example, miR-27a, as an inhibitor of MAPK as well as JNK during dendritic cell-mediated accumulation of Tregs in vivo, accelerated tumor growth by inducing the accumulation of immune cells, indicating that miR-27a is a potential target for cancer therapy [81].
Exosomic miRNAs in the tumor microenvironment
Alongside with well-known pathways by which cells can communicate, considerable attention is now being given to the role of extracellular vesicles (EVs) which have been shown to contain nucleic acids in form of miRNAs [137, 138]. More importantly, the discovery that EVs can deliver miRNAs to target cells, raising the possibility that such exosomes could work as a novel category of intercellular communicators [137, 139, 140]. Some researchers demonstrated that exosome-associated miRNAs are involved in the metastatic potential of malignancy. For example, EVs derived from metastatic breast cancer cells were shown to be able to deliver miR-200 to non-metastatic cells [141]. As outlined in a recent review [142], the tumor cells can “educate” the surrounding environment from normal to pro-inflammatory and pro-tumorigenic through exosome-dependent signals other than well-known cross talk.
Future directions and concluding remarks
Inflammatory circuits can differ considerably between different tumors in terms of cellular networks and cytokines. Understanding the diversity of the inflammatory microenvironment is instrumental to the design of therapeutic approaches targeting this microenvironment. Many anti-inflammatory drugs inhibit the activity of cytokines and tumor development in preclinical and clinical settings associated with cancer [143–145]. These conventional therapies mainly depend on genetic alterations of cancer cells and target cancer-associated inflammatory cell receptors or cytokines and their receptors [146].
Inflammatory cytokines, including TGF-β, play important roles in the regulation of tumor progression. Owing to the pleiotropic effects of TGF-β on the tumor microenvironment, targeting TGF-β signaling to directly treat cancer progression remains controversial. As described in this review, the TGF-β signaling pathway plays a critical role in the regulation of miRNA biogenesis. Given that a single miRNA can modulate the expression of hundreds of target genes, the regulation of miRNAs by the TGF-β signaling pathway could be critical for the modulation of the tumor microenvironment.
Several studies have investigated the role of TGF-β-related miRNAs in cancer and revealed the different mechanisms underlying the interaction between TGF-β and miRNAs in cancer. In TGF-β overexpressing cancer tissues, upregulated miRNAs may play a tumor-promoting role by downregulating the expression of tumor-suppressor genes. Therefore, downregulating the expression of these miRNAs and/or upregulating the expression of target genes could be a strategy for the treatment of cancer. On the other hand, the downregulation or inhibition of certain miRNAs in cancer may lead to oncogene overexpression and tumor-promoting effects. In this case, increasing the expression of these miRNAs and/or downregulating the expression of target genes could be applied for the treatment of cancer. In brief, understanding the molecular and epigenetic mechanisms underlying the relationship between inflammatory cytokine signaling and miRNAs in cancer may facilitate the development of new therapeutic strategies targeting the tumor microenvironment.
References
He L, Hannon GJ. MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet. 2004;5:522–31.
Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–97.
Melo SA, Esteller M. Dysregulation of microRNAs in cancer: playing with fire. FEBS Lett. 2010;585:2087–99.
Volinia S, Calin GA, Liu CG, Ambs S, Cimmino A, Petrocca F, et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci U S A. 2006;103:2257–61.
Ventura A, Jacks T. MicroRNAs and cancer: short RNAs go a long way. Cell. 2009;136:586–91.
Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer. 2006;6:857–66.
Kloosterman WP, Plasterk RHA. The diverse functions of microRNAs in animal development and disease. Dev Cell. 2006;11:441–50.
Marques-Rocha JL, Samblas M, Milagro FI, Bressan J, Martinez JA, Marti A. Noncoding RNAs, cytokines, and inflammation-related diseases. FASEB J Off Publ Fed Am Soc Exp Biol. 2015;29:3595–611.
O'Connell RM, Rao DS, Baltimore D. MicroRNA regulation of inflammatory responses. Annu Rev Immunol. 2012;30:295–312.
Mendell JT, Olson EN. MicroRNAs in stress signaling and human disease. Cell. 2012;148:1172–87.
Tili E, Michaille JJ, Calin GA. Expression and function of micro-RNAs in immune cells during normal or disease state. Int J Med Sci. 2008;5:73–9.
Sonkoly E, Stahle M, Pivarcsi A. MicroRNAs: novel regulators in skin inflammation. Clin Exp Dermatol. 2008;33:312–5.
Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454:436–44.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74.
Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001;357:539–45.
Facciabene A, Motz GT, Coukos G. T-regulatory cells: key players in tumor immune escape and angiogenesis. Cancer Res. 2012;72:2162–71.
Zhou X, Tang J, Cao H, Fan H, Li B: Tissue resident regulatory t cells: novel therapeutic targets for human disease. Cell Mol Immunol 2015
Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883–99.
Candido J, Hagemann T. Cancer-related inflammation. J Clin Immunol. 2013;33 Suppl 1:S79–84.
Tili E, Michaille JJ. Resveratrol, microRNAs, inflammation, and cancer. J Nucleic Acids. 2011;2011:102431.
Iorio MV, Croce CM. MicroRNA involvement in human cancer. Carcinogenesis. 2012;33:1126–33.
O'Neill LA, Sheedy FJ, McCoy CE. MicroRNAs: the fine-tuners of toll-like receptor signalling. Nat Rev Immunol. 2011;11:163–75.
Butz H, Rácz K, Hunyady L, Patócs A. Crosstalk between tgf-β signaling and the microRNA machinery. Trends Pharmacol Sci. 2012;33:382–93.
Sivadas VP, Kannan S. The microRNA networks of tgfbeta signaling in cancer. Tumour Biol. 2014;35:2857–69.
Tili E, Croce CM, Michaille JJ. Mir-155: on the crosstalk between inflammation and cancer. Int Rev Immunol. 2009;28:264–84.
Jurkovicova D, Magyerkova M, Kulcsar L, Krivjanska M, Krivjansky V, Gibadulinova A, et al. Mir-155 as a diagnostic and prognostic marker in hematological and solid malignancies. Neoplasma. 2014;61:241–51.
Tili E, Michaille JJ, Cimino A, Costinean S, Dumitru CD, Adair B, et al. Modulation of mir-155 and mir-125b levels following lipopolysaccharide/tnf-alpha stimulation and their possible roles in regulating the response to endotoxin shock. J Immunol. 2007;179:5082–9.
Xue H, Hua LM, Guo M, Luo JM. Ship1 is targeted by mir-155 in acute myeloid leukemia. Oncol Rep. 2014;32:2253–9.
Trotta R, Chen L, Ciarlariello D, Josyula S, Mao C, Costinean S, et al. Mir-155 regulates ifn-gamma production in natural killer cells. Blood. 2012;119:3478–85.
Yamada A, Horimatsu T, Okugawa Y, Nishida N, Honjo H, Ida H, et al. Serum mir-21, mir-29a and mir-125b are promising biomarkers for the early detection of colorectal neoplasia. Clin Cancer Res: Off J Am Assoc Cancer Res. 2015;21:4234–42.
Xu Z, Yu YQ, Ge YZ, Zhu JG, Zhu M, Zhao YC, et al. MicroRNA expression profiles in muscle-invasive bladder cancer: identification of a four-microRNA signature associated with patient survival. Tumour Biol. 2015;36:8159–66.
Luo S, Wang J, Ma Y, Yao Z, Pan H. Ppargamma inhibits ovarian cancer cells proliferation through upregulation of mir-125b. Biochem Biophys Res Commun. 2015;462:85–90.
Morelli E, Leone E, Cantafio ME, Di Martino MT, Amodio N, Biamonte L, et al. Selective targeting of irf4 by synthetic microrna-125b-5p mimics induces anti-multiple myeloma activity in vitro and in vivo. Leukemia. 2015. doi:10.1038/leu.2015.124.
Sheedy FJ, Palsson-McDermott E, Hennessy EJ, Martin C, O'Leary JJ, Ruan Q, et al. Negative regulation of tlr4 via targeting of the proinflammatory tumor suppressor pdcd4 by the microRNA mir-21. Nat Immunol. 2010;11:141–7.
Iliopoulos D, Hirsch HA, Struhl K. An epigenetic switch involving nf-kappab, lin28, let-7 microRNA, and il6 links inflammation to cell transformation. Cell. 2009;139:693–706.
Buscaglia LE, Li Y. Apoptosis and the target genes of microRNA-21. Chinese J Cancer. 2011;30:371–80.
Pfeffer SR, Yang CH, Pfeffer LM. The role of mir-21 in cancer. Drug Deve Res. 2015. doi:10.1002/ddr.21257.
Pedersen IM, Cheng G, Wieland S, Volinia S, Croce CM, Chisari FV, et al. Interferon modulation of cellular microRNAs as an antiviral mechanism. Nature. 2007;449:919–22.
Chen C, Zhang Y, Zhang L, Weakley SM, Yao Q. MicroRNA-196: critical roles and clinical applications in development and cancer. J Cell Mol Med. 2011;15:14–23.
Brest P, Lapaquette P, Souidi M, Lebrigand K, Cesaro A, Vouret-Craviari V, et al. A synonymous variant in irgm alters a binding site for mir-196 and causes deregulation of irgm-dependent xenophagy in crohn's disease. Nat Genet. 2011;43:242–5.
Yang G, Han D, Chen X, Zhang D, Wang L, Shi C, et al. Mir-196a exerts its oncogenic effect in glioblastoma multiforme by inhibition of ikappabalpha both in vitro and in vivo. Neuro-Oncology. 2014;16:652–61.
Qi J, Qiao Y, Wang P, Li S, Zhao W, Gao C. MicroRNA-210 negatively regulates lps-induced production of proinflammatory cytokines by targeting nf-kappab1 in murine macrophages. FEBS Lett. 2012;586:1201–7.
Dang K, Myers KA. The role of hypoxia-induced mir-210 in cancer progression. Int J Mol Sci. 2015;16:6353–72.
Wang J, Zhao J, Shi M, Ding Y, Sun H, Yuan F, et al. Elevated expression of mir-210 predicts poor survival of cancer patients: a systematic review and meta-analysis. PLoS One. 2014;9:e89223.
Chen WX, Ren LH, Shi RH. Implication of miRNAs for inflammatory bowel disease treatment: systematic review. World J Gastrointest Pathophysiol. 2014;5:63–70.
Ebrahimi F, Gopalan V, Smith RA, Lam AK. Mir-126 in human cancers: clinical roles and current perspectives. Exp Mol Pathol. 2014;96:98–107.
Chen H, Li L, Wang S, Lei Y, Ge Q, Lv N, et al. Reduced mir-126 expression facilitates angiogenesis of gastric cancer through its regulation on vegf-a. Oncotarget. 2014;5:11873–85.
Li D, Wang A, Liu X, Meisgen F, Grunler J, Botusan IR, et al. MicroRNA-132 enhances transition from inflammation to proliferation during wound healing. J Clin Invest. 2015;125:3008–26.
Wang ZH, Zhang QS, Duan YL, Zhang JL, Li GF, Zheng DL. Tgf-beta induced mir-132 enhances the activation of tgf-beta signaling through inhibiting smad7 expression in glioma cells. Biochem Biophys Res Commun. 2015;463:187–92.
Labbaye C, Testa U. The emerging role of mir-146a in the control of hematopoiesis, immune function and cancer. J Hematol Oncol. 2012;5:13.
Li J, Yang H, Li Y, Liu Y, Chen S, Qi C, et al. MicroRNA-146 up-regulation predicts the prognosis of non-small cell lung cancer by miRNA in situ hybridization. Exp Mol Pathol. 2014;96:195–9.
Davis-Dusenbery BN, Hata A. Mechanisms of control of microRNA biogenesis. J Biochem. 2010;148:381–92.
Costinean S, Zanesi N, Pekarsky Y, Tili E, Volinia S, Heerema N, et al. Pre-b cell proliferation and lymphoblastic leukemia/high-grade lymphoma in e(mu)-mir155 transgenic mice. Proc Natl Acad Sci U S A. 2006;103:7024–9.
Cremer TJ, Ravneberg DH, Clay CD, Piper-Hunter MG, Marsh CB, Elton TS, et al. Mir-155 induction by f. Novicida but not the virulent f. Tularensis results in ship down-regulation and enhanced pro-inflammatory cytokine response. PLoS One. 2009;4:e8508.
Bala S, Marcos M, Kodys K, Csak T, Catalano D, Mandrekar P, et al. Up-regulation of microRNA-155 in macrophages contributes to increased tumor necrosis factor {alpha} (tnf{alpha}) production via increased mRNA half-life in alcoholic liver disease. J Biol Chem. 2011;286:1436–44.
Roberts AB, Wakefield LM. The two faces of transforming growth factor beta in carcinogenesis. Proc Natl Acad Sci U S A. 2003;100:8621–3.
Akhurst RJ, Derynck R. Tgf-beta signaling in cancer--a double-edged sword. Trends Cell Biol. 2001;11:S44–51.
Blahna MT, Hata A. Smad-mediated regulation of microRNA biosynthesis. FEBS Lett. 2012;586:1906–12.
Hata A, Davis BN. Control of microRNA biogenesis by tgfbeta signaling pathway-a novel role of smads in the nucleus. Cytokine Growth Factor Rev. 2009;20:517–21.
Davis BN, Hata A. Regulation of microRNA biogenesis: a miriad of mechanisms: cell communication and signaling. CCS. 2009;7:18.
Elston R, Inman GJ. Crosstalk between p53 and tgf-beta signalling. J Signal Transduct. 2012;2012:294097.
Davis BN, Hilyard AC, Lagna G, Hata A. Smad proteins control drosha-mediated microRNA maturation. Nature. 2008;454:56–61.
Heldin CH, Moustakas A. Role of smads in tgfbeta signaling. Cell Tissue Res. 2011;347:21–36.
Davis BN, Hilyard AC, Nguyen PH, Lagna G, Hata A. Smad proteins bind a conserved RNA sequence to promote microRNA maturation by drosha. Mol Cell. 2010;39:373–84.
Wang W, Li J, Zhu W, Gao C, Jiang R, Li W, et al. MicroRNA-21 and the clinical outcomes of various carcinomas: a systematic review and meta-analysis. BMC Cancer. 2014;14:819.
Yu Y, Wang Y, Ren X, Tsuyada A, Li A, Liu LJ, et al. Context-dependent bidirectional regulation of the muts homolog 2 by transforming growth factor beta contributes to chemoresistance in breast cancer cells. Mol Cancer Res. 2010;8:1633–42.
Monfared H, Ziaee SA, Hashemitabar M, Khayatzadeh H, Kheyrollahi V, Tavallaei M, et al. Co-regulated expression of tgf-beta variants and mir-21 in bladder cancer. Urol J. 2013;10:981–7.
Hong L, Han Y, Zhang Y, Zhang H, Zhao Q, Wu K, et al. MicroRNA-21: a therapeutic target for reversing drug resistance in cancer. Expert Opin Ther Targets. 2013;17:1073–80.
Wang J, Li Y, Wang X, Jiang C. Ursolic acid inhibits proliferation and induces apoptosis in human glioblastoma cell lines u251 by suppressing tgf-beta1/mir-21/pdcd4 pathway. Basic Clin Pharmacol Toxicol. 2012;111:106–12.
Wang B, Hsu SH, Majumder S, Kutay H, Huang W, Jacob ST, et al. Tgfbeta-mediated upregulation of hepatic mir-181b promotes hepatocarcinogenesis by targeting timp3. Oncogene. 2010;29:1787–97.
Brockhausen J, Tay SS, Grzelak CA, Bertolino P, Bowen DG, d'Avigdor WM, et al. Mir-181a mediates tgf-beta-induced hepatocyte emt and is dysregulated in cirrhosis and hepatocellular cancer. Liver Int. 2014;35:240–53.
Wang Y, Yu Y, Tsuyada A, Ren X, Wu X, Stubblefield K, et al. Transforming growth factor-beta regulates the sphere-initiating stem cell-like feature in breast cancer through miRNA-181 and atm. Oncogene. 2011;30:1470–80.
Neel JC, Lebrun JJ. Activin and tgfbeta regulate expression of the microRNA-181 family to promote cell migration and invasion in breast cancer cells. Cell Signal. 2013;25:1556–66.
Liu Y, Lai L, Chen Q, Song Y, Xu S, Ma F, et al. MicroRNA-494 is required for the accumulation and functions of tumor-expanded myeloid-derived suppressor cells via targeting of pten. J Immunol. 2012;188:5500–10.
Li L, Li Z, Kong X, Xie D, Jia Z, Jiang W, et al. Down-regulation of microRNA-494 via loss of smad4 increases foxm1 and beta-catenin signaling in pancreatic ductal adenocarcinoma cells. Gastroenterology. 2014;147:485–97. e418.
Ma L, Teruya-Feldstein J, Weinberg RA. Tumour invasion and metastasis initiated by microRNA-10b in breast cancer. Nature. 2007;449:682–8.
Ma L, Reinhardt F, Pan E, Soutschek J, Bhat B, Marcusson EG, et al. Therapeutic silencing of mir-10b inhibits metastasis in a mouse mammary tumor model. Nat Biotechnol. 2010;28:341–7.
Ma L. Role of mir-10b in breast cancer metastasis. Breast Cancer Res : BCR. 2010;12:210.
Han X, Yan S, Weijie Z, Feng W, Liuxing W, Mengquan L, et al. Critical role of mir-10b in transforming growth factor-beta1-induced epithelial-mesenchymal transition in breast cancer. Cancer Gene Ther. 2014;21:60–7.
Ouyang H, Gore J, Deitz S, Korc M. MicroRNA-10b enhances pancreatic cancer cell invasion by suppressing tip30 expression and promoting egf and tgf-beta actions. Oncogene. 2014;33:4664–74.
Min S, Li L, Zhang M, Zhang Y, Liang X, Xie Y, et al. Tgf-beta-associated mir-27a inhibits dendritic cell-mediated differentiation of th1 and th17 cells by tab3, p38 mapk, map2k4 and map2k7. Genes Immun. 2012;13:621–31.
Donatelli SS, Zhou JM, Gilvary DL, Eksioglu EA, Chen X, Cress WD, et al. Tgf-beta-inducible microRNA-183 silences tumor-associated natural killer cells. Proc Natl Acad Sci U S A. 2014;111:4203–8.
Song L, Liu L, Wu Z, Li Y, Ying Z, Lin C, et al. Tgf-beta induces mir-182 to sustain nf-kappab activation in glioma subsets. J Clin Invest. 2012;122:3563–78.
Kong W, Yang H, He L, Zhao JJ, Coppola D, Dalton WS, et al. MicroRNA-155 is regulated by the transforming growth factor beta/smad pathway and contributes to epithelial cell plasticity by targeting rhoa. Mol Cell Biol. 2008;28:6773–84.
Gal H, Pandi G, Kanner AA, Ram Z, Lithwick-Yanai G, Amariglio N, et al. Mir-451 and imatinib mesylate inhibit tumor growth of glioblastoma stem cells. Biochem Biophys Res Commun. 2008;376:86–90.
Kumar MS, Lu J, Mercer KL, Golub TR, Jacks T. Impaired microRNA processing enhances cellular transformation and tumorigenesis. Nat Genet. 2007;39:673–7.
Schoof CR, Botelho EL, Izzotti A, Vasques Ldos R. MicroRNAs in cancer treatment and prognosis. Am J Cancer Res. 2012;2:414–33.
Gregory PA, Bert AG, Paterson EL, Barry SC, Tsykin A, Farshid G, et al. The mir-200 family and mir-205 regulate epithelial to mesenchymal transition by targeting zeb1 and sip1. Nat Cell Biol. 2008;10:593–601.
Truong HH, Xiong J, Ghotra VP, Nirmala E, Haazen L, Le Devedec SE, et al. Beta1 integrin inhibition elicits a prometastatic switch through the tgfbeta-mir-200-zeb network in e-cadherin-positive triple-negative breast cancer. Sci Signal. 2014;7:ra15.
Izumchenko EG, Chang X, Michailidi C, Kagohara LT, Ravi R, Paz K, et al. The tgfbeta-mir200-mig6 pathway orchestrates the emt-associated kinase switch that induces resistance to egfr inhibitors. Cancer Res. 2014;74:3995–4005.
Maroof H, Salajegheh A, Smith RA, Lam AK. MicroRNA-34 family, mechanisms of action in cancer: a review. Curr Cancer Drug Targets. 2014;14:737–51.
Dimopoulos K, Gimsing P, Gronbaek K. Aberrant microRNA expression in multiple myeloma. Eur J Haematol. 2013;91:95–105.
Yang P, Li Q-J, Feng Y, Zhang Y, Markowitz G, Ning S, et al. Tgf-β-mir-34a-ccl22 signaling-induced treg cell recruitment promotes venous metastases of hbv-positive hepatocellular carcinoma. Cancer Cell. 2012;22:291–303.
Sonkoly E, Wei T, Pavez Lorie E, Suzuki H, Kato M, Torma H, et al. Protein kinase c-dependent upregulation of mir-203 induces the differentiation of human keratinocytes. J Invest Dermatol. 2010;130:124–34.
Tian L, Li M, Ge J, Guo Y, Sun Y, Liu M, et al. Mir-203 is downregulated in laryngeal squamous cell carcinoma and can suppress proliferation and induce apoptosis of tumours. Tumour Biol. 2014;35:5953–63.
Xu M, Gu M, Zhang K, Zhou J, Wang Z, Da J. Mir-203 inhibition of renal cancer cell proliferation, migration and invasion by targeting of fgf2. Diagn Pathol. 2015;10:24.
Ding X, Park SI, McCauley LK, Wang CY. Signaling between transforming growth factor beta (tgf-beta) and transcription factor snai2 represses expression of microRNA mir-203 to promote epithelial-mesenchymal transition and tumor metastasis. J Biol Chem. 2013;288:10241–53.
Ueno K, Hirata H, Shahryari V, Chen Y, Zaman MS, Singh K, et al. Tumour suppressor microRNA-584 directly targets oncogene rock-1 and decreases invasion ability in human clear cell renal cell carcinoma. Br J Cancer. 2011;104:308–15.
Fils-Aime N, Dai M, Guo J, El-Mousawi M, Kahramangil B, Neel JC, et al. MicroRNA-584 and the protein phosphatase and actin regulator 1 (phactr1), a new signaling route through which transforming growth factor-beta mediates the migration and actin dynamics of breast cancer cells. J Biol Chem. 2013;288:11807–23.
Sun MM, Li JF, Guo LL, Xiao HT, Dong L, Wang F, et al. Tgf-[beta]1 suppression of microRNA-450b-5p expression: a novel mechanism for blocking myogenic differentiation of rhabdomyosarcoma. Oncogene. 2014;33:2075–86.
Martin J, Jenkins RH, Bennagi R, Krupa A, Phillips AO, Bowen T, et al. Post-transcriptional regulation of transforming growth factor beta-1 by microRNA-744. PLoS One. 2011;6:e25044.
Dogar AM, Towbin H, Hall J. Suppression of latent transforming growth factor (tgf)-beta1 restores growth inhibitory tgf-beta signaling through microRNAs. J Biol Chem. 2011;286:16447–58.
Dogar AM, Semplicio G, Guennewig B, Hall J. Multiple microRNAs derived from chemically synthesized precursors regulate thrombospondin 1 expression. Nucleic Acid Ther. 2014;24:149–59.
Braun J, Hoang-Vu C, Dralle H, Huttelmaier S. Downregulation of microRNAs directs the emt and invasive potential of anaplastic thyroid carcinomas. Oncogene. 2010;29:4237–44.
Masri S, Liu Z, Phung S, Wang E, Yuan YC, Chen S. The role of microRNA-128a in regulating tgfbeta signaling in letrozole-resistant breast cancer cells. Breast Cancer Res Treat. 2010;124:89–99.
Jiang X, Xiang G, Wang Y, Zhang L, Yang X, Cao L, et al. MicroRNA-590-5p regulates proliferation and invasion in human hepatocellular carcinoma cells by targeting tgf-beta rii. Mol Cells. 2012;33:545–51.
Feng B, Dong TT, Wang LL, Zhou HM, Zhao HC, Dong F, et al. Colorectal cancer migration and invasion initiated by microRNA-106a. PLoS One. 2012;7:e43452.
Lo SS, Hung PS, Chen JH, Tu HF, Fang WL, Chen CY, et al. Overexpression of mir-370 and downregulation of its novel target tgfbeta-rii contribute to the progression of gastric carcinoma. Oncogene. 2011;31:226–37.
Chu TH, Yang CC, Liu CJ, Lui MT, Lin SC, Chang KW. Mir-211 promotes the progression of head and neck carcinomas by targeting tgfbetarii. Cancer Lett. 2013;337:115–24.
Yu Y, Kanwar SS, Patel BB, Oh P-S, Nautiyal J, Sarkar FH, et al. MicroRNA-21 induces stemness by downregulating transforming growth factor beta receptor 2 (ttgf-β r2) in colon cancer cells. Carcinogenesis. 2012;33:68–76.
Mishra S, Deng JJ, Gowda PS, Rao MK, Lin CL, Chen CL, et al. Androgen receptor and microRNA-21 axis downregulates transforming growth factor beta receptor ii (tgfbr2) expression in prostate cancer. Oncogene. 2014;33:4097–106.
Keklikoglou I, Koerner C, Schmidt C, Zhang JD, Heckmann D, Shavinskaya A, et al. MicroRNA-520/373 family functions as a tumor suppressor in estrogen receptor negative breast cancer by targeting nf-kappab and tgf-beta signaling pathways. Oncogene. 2011;31:4150–63.
Harazono Y, Muramatsu T, Endo H, Uzawa N, Kawano T, Harada K, et al. Mir-655 is an emt-suppressive microRNA targeting zeb1 and tgfbr2. PLoS One. 2013;8:e62757.
Jiang Z, Yin J, Fu W, Mo Y, Pan Y, Dai L, et al. MiRNA 17 family regulates cisplatin-resistant and metastasis by targeting tgfbetar2 in nsclc. PLoS One. 2014;9:e94639.
Jiang F, Mu J, Wang X, Ye X, Si L, Ning S, et al. The repressive effect of mir-148a on tgf beta-smads signal pathway is involved in the glabridin-induced inhibition of the cancer stem cells-like properties in hepatocellular carcinoma cells. PLoS One. 2014;9:e96698.
Turcatel G, Rubin N, El-Hashash A, Warburton D. Mir-99a and mir-99b modulate tgf-β induced epithelial to mesenchymal plasticity in normal murine mammary gland cells. PLoS One. 2012;7:e31032.
Wu ZB, Cai L, Lin SJ, Lu JL, Yao Y, Zhou LF. The mir-92b functions as a potential oncogene by targeting on smad3 in glioblastomas. Brain Res. 2013;1529:16–25.
Geraldo MV, Yamashita AS, Kimura ET. MicroRNA mir-146b-5p regulates signal transduction of tgf-beta by repressing smad4 in thyroid cancer. Oncogene. 2012;31:1910–22.
Zhang Y, Fan K-J, Sun Q, Chen A-Z, Shen W-L, Zhao Z-H, et al. Functional screening for miRNAs targeting smad4 identified mir-199a as a negative regulator of tgf-β signalling pathway. Nucleic Acids Res. 2012;40:9286–97.
Hager M, Pedersen CC, Larsen MT, Andersen MK, Hother C, Gronbaek K, et al. MicroRNA-130a-mediated down-regulation of smad4 contributes to reduced sensitivity to tgf-beta1 stimulation in granulocytic precursors. Blood. 2011;118:6649–59.
Liu L, Nie J, Chen L, Dong G, Du X, Wu X, et al. The oncogenic role of microRNA-130a/301a/454 in human colorectal cancer via targeting smad4 expression. PLoS One. 2013;8:e55532.
Genovese G, Ergun A, Shukla SA, Campos B, Hanna J, Ghosh P, et al. MicroRNA regulatory network inference identifies mir-34a as a novel regulator of tgf-β signaling in glioblastoma. Cancer Disc. 2012;2:736–49.
Zhong H, Wang HR, Yang S, Zhong JH, Wang T, Wang C, et al. Targeting smad4 links microRNA-146a to the tgf-beta pathway during retinoid acid induction in acute promyelocytic leukemia cell line. Int J Hematol. 2010;92:129–35.
Rai D, Kim SW, McKeller MR, Dahia PL, Aguiar RC. Targeting of smad5 links microRNA-155 to the tgf-beta pathway and lymphomagenesis. Proc Natl Acad Sci U S A. 2010;107:3111–6.
Jiang D, Aguiar RC. MicroRNA-155 controls rb phosphorylation in normal and malignant b lymphocytes via the noncanonical tgf-beta1/smad5 signaling module. Blood. 2014;123:86–93.
Liu Y, Sun R, Lin X, Liang D, Deng Q, Lan K. Kaposi's sarcoma-associated herpesvirus-encoded microRNA mir-k12-11 attenuates transforming growth factor beta signaling through suppression of smad5. J Virol. 2012;86:1372–81.
Smith AL, Iwanaga R, Drasin DJ, Micalizzi DS, Vartuli RL, Tan AC, et al. The mir-106b-25 cluster targets smad7, activates tgf-beta signaling, and induces emt and tumor initiating cell characteristics downstream of six1 in human breast cancer. Oncogene. 2012;31:5162–71.
Xia H, Ooi LL, Hui KM. MicroRNA-216a/217-induced epithelial-mesenchymal transition targets pten and smad7 to promote drug resistance and recurrence of liver cancer. Hepatology. 2013;58:629–41.
Petrocca F, Vecchione A, Croce CM. Emerging role of mir-106b-25/mir-17-92 clusters in the control of transforming growth factor beta signaling. Cancer Res. 2008;68:8191–4.
Petrocca F, Visone R, Onelli MR, Shah MH, Nicoloso MS, de Martino I, et al. E2f1-regulated microRNAs impair tgfbeta-dependent cell-cycle arrest and apoptosis in gastric cancer. Cancer Cell. 2008;13:272–86.
Mestdagh P, Bostrom AK, Impens F, Fredlund E, Van Peer G, De Antonellis P, et al. The mir-17-92 microRNA cluster regulates multiple components of the tgf-beta pathway in neuroblastoma. Mol Cell. 2010;40:762–73.
Li J, Fu H, Xu C, Tie Y, Xing R, Zhu J, et al. Mir-183 inhibits tgf-beta1-induced apoptosis by downregulation of pdcd4 expression in human hepatocellular carcinoma cells. BMC Cancer. 2010;10:354.
Pollari S, Leivonen SK, Perala M, Fey V, Kakonen SM, Kallioniemi O. Identification of microRNAs inhibiting tgf-beta-induced il-11 production in bone metastatic breast cancer cells. PLoS One. 2012;7:e37361.
Jiang H, Jin C, Liu J, Hua D, Zhou F, Lou X, et al. Next generation sequencing analysis of miRNAs: miR-127-3p inhibits glioblastoma proliferation and activates tgf-beta signaling by targeting ski. OMICS. 2014;18:196–206.
Zhang YE. Non-smad pathways in tgf-beta signaling. Cell Res. 2009;19:128–39.
Mu Y, Gudey SK, Landstrom M. Non-smad signaling pathways. Cell Tissue Res. 2012;347:11–20.
Sato-Kuwabara Y, Melo SA, Soares FA, Calin GA. The fusion of two worlds: non-coding RNAs and extracellular vesicles—diagnostic and therapeutic implications (review). Int J Oncol. 2015;46:17–27.
Raposo G, Stoorvogel W. Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol. 2013;200:373–83.
Villarroya-Beltri C, Baixauli F, Gutierrez-Vazquez C, Sanchez-Madrid F, Mittelbrunn M. Sorting it out: regulation of exosome loading. Semin Cancer Biol. 2014;28:3–13.
Squadrito ML, Baer C, Burdet F, Maderna C, Gilfillan GD, Lyle R, et al. Endogenous RNAs modulate microRNA sorting to exosomes and transfer to acceptor cells. Cell Rep. 2014;8:1432–46.
Le MT, Hamar P, Guo C, Basar E, Perdigao-Henriques R, Balaj L, et al. Mir-200-containing extracellular vesicles promote breast cancer cell metastasis. J Clin Invest. 2014;124:5109–28.
Neviani P, Fabbri M. Exosomic microRNAs in the tumor microenvironment. Frontiers Med. 2015;2:47.
Eugenia CY: Interaction of salicylates and the other nonsteroidal anti-inflammatory agents with breast cancer endocrine treatment: Systematic review. Am J Clin Oncol 2014:in press.
Rothwell PM, Fowkes FG, Belch JF, Ogawa H, Warlow CP, Meade TW. Effect of daily aspirin on long-term risk of death due to cancer: analysis of individual patient data from randomised trials. Lancet. 2011;377:31–41.
Shi L, Wang L, Hou J, Zhu B, Min Z, Zhang M, et al. Targeting roles of inflammatory microenvironment in lung cancer and metastasis. Cancer Metastasis Rev. 2015;34:319–31.
Balkwill FR, Mantovani A. Cancer-related inflammation: common themes and therapeutic opportunities. Semin Cancer Biol. 2012;22:33–40.
Acknowledgments
The authors would like to acknowledge funding from Chinese Nature Science Foundation (81272738, 81372867, 81472297) and project founded by the Priority Academic Program Development of Jiangsu Higher Education Institutions.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
None
Additional information
Lingling Guo, Yongsheng Zhang and Lifeng Zhang contributed equally to this work.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Guo, L., Zhang, Y., Zhang, L. et al. MicroRNAs, TGF-β signaling, and the inflammatory microenvironment in cancer. Tumor Biol. 37, 115–125 (2016). https://doi.org/10.1007/s13277-015-4374-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13277-015-4374-2