
Abstract
Tumor-associated myeloid cells have been implicated in regulating many of the “hallmarks of cancer” and thus fostering solid tumor development and metastasis. However, the same innate leukocytes also participate in anti-tumor immunity and restraint of malignant disease. While many factors regulate the propensity of myeloid cells to promote or repress cancerous growths, polarized adaptive immune responses by B and T lymphocytes have been identified as regulators of many aspects of myeloid cell biology by specifically regulating their functional capabilities. Here, we detail the diversity of heterogeneous B and T lymphocyte populations and their impacts on solid tumor development through their abilities to regulate myeloid cell function in solid tumors.
Similar content being viewed by others


1 Introduction
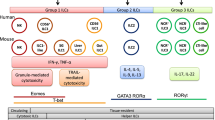
Virchow first described leukocyte infiltration of solid tumors in the 1800s; however, only recently have we begun to understand the diverse regulatory roles played by immune cells during cancer development. Historically, leukocytes found in and around developing tumors were thought to represent an attempt by the host to eradicate neoplastic cells. Indeed, some leukocytes, including CD8+ cytotoxic T lymphocytes (CTLs) and natural killer (NK) cells, do play a critical role in restraining tumor development [1]. However, we now appreciate that the significance of these anti-tumor programs can be thwarted by other subsets of leukocytes that instead foster tumor development [2–5]. Immune-competent mouse models of human cancer have enabled a detailed evaluation of the tumor-promoting capacity of several subsets of myeloid cells, including mast cells (MCs), monocytes, granulocytes/neutrophils, and macrophages, as well as some subsets of lymphocytes [6, 7]. However, depending on their differentiation status and immune microenvironment, subpopulations of these same cells can also support tumor rejection and response to anti-cancer therapy [2, 8, 9], thus indicating that pro- and anti-tumor programming of leukocytes is dynamic. In this review, we discuss recent insights into the role of B and T lymphocytes as “gatekeepers” of myeloid cell bioactivity (Fig. 1) and how recognition of these dynamic interactions reveals novel opportunities for anti-cancer therapy.

Adaptive immune responses control tumor-associated myeloid cell bioactivity and tumor progression. Polarized responses by adaptive immune cells alter the balance between pro- and anti-tumor myeloid cell bioactivities. When the host’s response to neoplastic cell growth results in the production of TH1 cytokines by CD4+ T lymphocytes and NK cells, myeloid cells in turn induce programs promoting tumor regression and/or dormant disease. However, when these adaptive immune responses include chronic B lymphocyte activation and IgG production in combination with TH2 and TREG lymphocyte activation, programs of immune suppression, angiogenesis, tissue remodeling, and invasion are favored in myeloid cells and contribute to tumor progression and metastasis
2 Paradoxical role of CD4+ T lymphocytes in solid tumor development
In contrast to CD8+ CTLs that play well-defined roles in hindering cancer development, the functional significance of CD4+ T lymphocytes in tumor progression appears more paradoxical. For example, retrospective evaluation of colon and lung carcinomas revealed that extensive infiltration of tumors by CD4+ T cells correlates with favorable clinical outcome, whereas in breast and renal cancers exhibiting similar infiltrations instead correlates with decreased overall survival [10–13]. Analysis of mouse models of human cancer have provided some clarity for these disparate findings and revealed that etiology and organ specificity matters with regards to how CD4+ T cells aid or constrain tumor progression. Schreiber and colleagues demonstrated that, whereas CD4+ T cell deficiency enhances methylcholanthrene (MCA)-initiated sarcoma development [14], carcinoma development on the other hand is inhibited following two-stage squamous carcinogenesis [15, 16]. Similarly, in a mouse model of skin and cervical carcinoma development where oncogenes from human papilloma virus type 16 (HPV16) are expressed under the control of the keratin 14 promoter, skin carcinoma formation is modestly attenuated by CD4+ T cell deficiency, whereas cervical carcinoma development is significantly enhanced [17, 18]. Together, these observations demonstrate that immune responses accompanying tumor development are organ dependent and, based on the neoplastic microenvironment, engage either pro- or anti-tumor immune programs. The heterogeneity of CD4+ T cell subsets that accumulate in tissues may be at the heart of these paradoxical findings.
3 CD4+ T lymphocyte heterogeneity
CD4+ T cells represent a highly heterogeneous population of cells that develop along different functional lineages depending upon polarizing cytokine signals during activation by antigens [19]. Classically, CD4+ T lymphocyte subsets include TH1 and TH2 lineages that are characteristically fostered by exposure to interleukin (IL)12 and IL4, respectively. Following activation, CD4+ T cells assuming the TH1 fate secrete interferon (IFN)γ, tumor necrosis factor (TNF)α, IL2, and IL12 [20]. Through the production of these cytokines, TH1 cells regulate immune surveillance programs by up-regulating antigen processing and presentation on major histocompatability complex (MHC) I and II molecules by professional antigen presenting cells (APCs) and as such can regulate the duration and magnitude of CD8+ CTL responses [21]. In addition, following strong antigen-specific activation, TH1 cells can directly kill tumor cells by releasing high levels of IFNγ, TNFα and cytolytic granules. Thus, when present, TH1 responses can directly and indirectly regulate anti-tumor programs that restrain cancer development.
In contrast, TH2 CD4+ T cells express high levels of IL4, -5, -6, -10, and -13 that, together, alter adaptive immunity by inducing T cell anergy, inhibiting T cell-mediated cytotoxicity as well as fostering humoral immune responses directed by B cells [22, 23]. The TH2 cytokines IL4 and IL13 are important mediators of CD4+ T cell functionality in vitro, TH2 CD4+ T cells inhibit apoptosis and induce proliferation of breast carcinoma cells; in vivo, IL4 emanating from CD4+ T cells fosters breast cancer growth [24, 25]. Consistent with these findings, a high ratio of TH2+ to TH1+ cells correlates with parameters of clinical disease progression, such as increased tumor size and grade and lymph node metastasis of breast cancers [26].
Adding to this TH1 versus TH2 paradigm, CD4 lineages have recently expanded to include a TH17 subset that is differentiated by a combination of IL6 and transforming growth factor (TGF)β and mediate their effects through secretion of IL17, -21, and -22, [27–29]. TH17 cells are thought to play an important role in protection against some extracellular pathogens and in regulating auto-immune disease [30, 31]. As such, TH17 cells have been implicated in the development of inflammation-associated colonic tumors in response to pathogenic bacteria [32]. TH17 cell infiltration has also been observed in patients with colon, ovarian, prostate, and hepatocellular carcinoma where high numbers of IL17-producing cells correlates with poor prognosis [33–35]. In mouse models of non-small cell lung cancer (NSCLC), IL17 enhanced tumor growth by promoting development of angiogenic vasculature [36, 37]. In contrast, in a B16 melanoma model, IL17 depletion rendered mice more susceptible to metastasis [38], a phenotype that was blocked by adoptive transfer of tumor-specific TH17 cells that fostered immune surveillance by CD8+ CTLs and dendritic cells (DCs) [38]. Together, these experimental findings indicate that the role of TH17 cells in regulating aspects of cancer development may also be context dependent.
In addition to TH1, TH2, and TH17 CD4+ effectors, populations of CD4+FoxP3+ T regulatory (TREG) cells are also thought to play a considerable role in regulating tumor immunity. In human cancers, increased prevalence of CD4+FoxP3+ TREG cells correlates with increased survival for follicular lymphoma [39], while it instead correlates with poor prognosis in pancreatic ductal carcinoma [40], non-small cell lung cancer [41], renal cell carcinoma [11], and breast carcinoma [42]. Suppression of the anti-tumor activities of CD8+ CTLs, NKs, and DCs is at the heart of how TREG cells control tumor development [43, 44]; however, the multitude of mechanisms whereby they regulate anti-tumor programs suggest the existence of distinct tissue-specific sub-populations of TREG cells, each endowed with or capable of various bioeffector activities [45–47]. Mechanistic studies have revealed that TREG cells support pro-tumor immunity not only by increasing local levels of immunosuppressive cytokines including TGFβ, IL35, and IL10 but also by direct cytolytic effects through production of perforin and granzyme. In addition, TREG cells can disrupt the metabolic activity of cyclic adenosine monophosphate (cAMP) transfer, as well as inhibit APC function by inducing binding of CTLA-4 to CD80/86 [44, 48].
Thus, while it is now clear that a spectrum of CD4+ T cell subtypes are present in human tumors of essentially all types, the role they play in promoting or repressing tumor development likely has to do with the type of CD4+ T cell subtype that is either recruited to or accumulates within each distinct tumor microenvironment. These in turn then regulate anti-tumor programs by professional cytotoxic cells (CD8+ T CTLs and NK cells), as well as regulating pro-tumor properties of a diverse array of myeloid cell subtypes as discussed below.
4 Myeloid heterogeneity and tumor development
Innate immune cells of myeloid origin, e.g., granulocytes (neutrophils, basophils, and eosinophils), DCs, macrophages, NK cells, and MCs, are also prominent components of pre- and malignant tissues and functionally contribute to cancer development by releasing a myriad of cytokines, chemokines, matrix metalloproteinases, serine proteases, DNA-damaging molecules (reactive oxygen species), histamine, and other bioactive mediators that regulate tissue remodelling and angiogenesis [49–53], suppress anti-tumor immunity [54–56], and enhance tumor cell survival, migration, and metastasis [57, 58].
Nucleated hematopoietic cells that have been directly implicated in tumor angiogenesis include MCs [51], tumor-associated macrophages (TAMs) [5, 23, 59], Tie2-expressing monocytes [50, 60], neutrophils [52], DC precursors [61], and myeloid immune suppressor cells [62, 63]. Other hematopoietic cell types, such as platelets [64], eosinophils [65], and hematopoietic progenitors [66], also participate in angiogenic processes, but it remains to be established whether they can directly promote tumor angiogenesis, rather than having a broader function in supporting tissue inflammation and remodelling.
In contrast, these same myeloid cell lineages also foster tumor rejection by inducing angiostatic programs, enhancing CTL and NK responses, and directly inducing tumor cell death [13]. As an example of these paradoxical roles, studies from several laboratories have reported that TAMs enhance angiogenesis and metastasis of malignant mammary tumors [25, 67, 68]. In contrast, TAMs exposed to toll-like receptor (TLR) ligands and/or IFNγ directly lyse mammary tumor cells, increase antigen presentation, and secrete angiostatic proteins such as CXCL10 and 11 [8, 9 , 69]. These distinct bioactivities are mirrored in neutrophils, MCs, and DCs and may be due to the inherent plasticity of myeloid lineage cells regulated by local factors present in distinct tissue and/or organ microenvironments.
The bioactive states of macrophages, as well as other myeloid cells, have been classified according to TH1 and TH2 nomenclature, referred to as M1 (classical) or M2 (alternative) activation, respectively [2, 70, 71] (Fig. 1). M1 macrophages are regulated by TH1 cytokines including IFNγ, TNFα, and granulocyte monocyte-colony stimulating factor (GM-CSF) that enhance macrophage cytotoxic activity, production of pro-inflammatory cytokines, and antigen presentation capacity [70, 71]. In contrast, tissue macrophages can achieve various alternatively activated M2 states following exposure to TH2 cytokines, including IL4 or IL13 (M2a), potentiation by immune complexes and TLR ligands (M2b) or immunosuppressive cytokines including IL10 or TGFβ and/or glucocorticoid hormones (M2c) [70].
The general hallmarks of M2 macrophages include high levels of IL10, IL1Ra, IL1 decoy receptor CCL17 and CCL22 secretion, high expression of mannose, scavenger and galactose-type receptors, low expression of IL12, as well as poor APC capability. Intriguingly, although these alternative activation states (M2a, b, and c) share many phenotypic characteristics, they are distinct and induce individual context-dependent environmental responses. For example, induction of an M2c phenotype by IL10 results in highly immune suppressive macrophages that can also produce matrix components such as versican or PTX3. In contrast, TH2 cytokine induction of M2a TAMs induces expression of fibronectin, as well as catabolism of L-arginine by arginase that in turn leads to increased collagen synthesis and matrix remodeling [70, 72]. Our own work has revealed that IL4 and/or IL13 activation of macrophages induces production of growth factors including epidermal growth factor, TGFβ, and basic fibroblast growth factor that together regulate invasive, angiogenic, and immune-suppressive programs [25] (unpublished data). Both M2a and M2b macrophages down-regulate the pro-inflammatory cytokines IL1, IL6, and TNFα [73], whereas induction of M2b macrophages by immune complexes induces these same inflammatory cytokines in addition to IL10 and also likely enhances vascular responses such as endothelial migration and vessel dilation [74]. Thus, M2-polarized cells promote scavenging of debris, angiogenesis, and remodeling and repair of wounded/damaged tissues. Parallel and non-redundant activity states have also been defined for DCs (i.e., DC1, DC2) [75] and neutrophils (i.e., N1, N2) [76] (Fig. 1).
To address pro- versus anti-tumor capabilities of these opposing states, Hagemann and colleagues demonstrated that by “reprogramming” M2 TAMs through deletion of IKKβ macrophage phenotype could be switched from immunosuppressive to actively promoting immune surveillance, as reflected by decreases in IL10 and arginase-1 expression and increased IL12 and nitric oxide production together resulting in decreased ovarian tumor growth through recruitment and activation of NK cells [67, 77]. Again, organ specificity and/or etiology may play a role in regulating how reprogramming can be achieved. In a mouse model of squamous carcinogenesis, we recently reported that FcγR signaling in myeloid cells directly regulates whether myeloid cells enhance or repress cancer development, which correlate with their unique gene expression signatures that reflect M1 versus M2 and DC1 versus DC2 programs [74]. These data indicate the significance of reprogramming myeloid cell phenotypes to affect tumor outcome. The major question that arises with regards to this capability then becomes what are the cellular and molecular programs in tissues and/or tumors that regulate the bioactive state of these important myeloid cells and how recognition of these can be translated into anti-cancer therapy.
5 T lymphocytes as regulators of anti-tumor macrophages
Establishment of an immune reaction in homeostatic tissue typically involves activation of NK cells in response to stress signals or infectious agents, whom by their production of IFNγ in turn prime macrophages towards an M1 state, culminating in enhanced presence of macrophages with cytotoxic capability [71]. However, production of IFNγ by NK cells is generally transient and therefore insufficient to sustain M1 macrophage polarization; thus, IFNγ-producing TH1 cells are critical for immune responses requiring sustained M1 macrophage bioactivities. In tumors, studies by Corthay and colleagues demonstrated that TH1 cell regulation of locally activated M1 macrophages were significant and fostered rejection of myeloma and lymphomas in the absence of CTL responses [69, 78]. Moreover, expression of IL12 and IFNγ by TH1 cells can combine to enhance anti-tumor responses by NK and NK-T cells by up-regulating expression of NK receptors such as NKG2D (in response to IL12) and expression of NK receptor ligands such as RAE1 on target cells (in response to IFNγ) [79]. TH1 responses then in turn favor anti-tumor NK and macrophage responses that eliminate neoplastic cells. However, while TH1 cells are antigen specific, tumoricidal macrophages exert indiscriminate cell killing activity. Thus, multiple immunosuppressive programs have evolved to eliminate the adverse autoimmune pathologies, such as rheumatoid arthritis, that are associated with over-activation of these M1 responses [80]; unfortunately, many of these immunosuppressive programs are usurped by developing cancers.
6 T lymphocytes as regulators of pro-tumor myeloid cells
In contrast to induction of tumor-immune surveillance programs by TH1 cells, TREG and TH2 cells have the capacity to induce alternative activation states of macrophages, DCs, and neutrophils involved in promoting cancer development. Studies of human TREGs have demonstrated their ability to block classical activation of macrophages and instead foster immunosuppressive myeloid phenotypes through the production of IL10 and TGFβ [81]. Similar biology may apply to neutrophils, as recently reported by Fridlender and colleagues who found that loss of TGFβ signaling through ALK4/5 inhibition resulted in recruitment of N1-polarized neutrophils with tumoricidal bioactivities [76]. While TGFβ in the tumor microenvironment is produced by multiple cell types, these data may indicate that TREG cells suppress N1 tumoricidal responses through production of TGFβ and, as such, favor pro-tumor N2 or immature monocyte (IMC) phenotypes. Our own work has demonstrated that CD4+ TH2 cells in mammary tumors promote M2 responses in TAM and IMCs that in turn enhance pulmonary metastasis [25]. Together, these data indicate that the balance of TH1 versus TH2/TREG responses regulates the pro- versus anti-tumor programming of tumor-associated myeloid cells.
7 B lymphocytes as regulators of myeloid cells during cancer development
B lymphocytes constitute a central component of humoral immunity and not only serve in antibody production but also in antigen presentation and cytokine secretion. In particular, B lymphocyte expression of MHC and co-stimulatory molecules as well as secretion of pro-inflammatory cytokines are critical for regulating CD4+ and CD8+ T cell activation, expansion, antigenic spreading, and memory T lymphocyte formation. The heterogeneity of B lymphocyte responses has been recently recognized and diverse B cell subtypes with either pro-immune or regulatory properties have been identified in vivo. Precisely, regulatory B lymphocytes (BREG), which include various subtypes of IL10-producing cells, have been identified in the context of autoimmune diseases and exert anti-inflammatory functions [82, 83]. However, a role for BREG cells in cancer has not been fully elucidated.
B lymphocytes in general have only recently gained recognition for representing significant components of tumor immunity [84]. B cell involvement in solid tumor development was initially described in syngeneic allograft murine tumor models in combination with genetic or antibody-mediated B cell depletion. In these studies, B cell-deficient mice (μMT) exhibited resistance to several types of syngeneic tumors, including EL4 thymoma, MC38 colon carcinoma, and B16 and D5 melanoma [85, 86], whereas partial B cell depletion resulted in significantly reduced tumor burden in a transplantable model of colorectal cancer [87]. A tumor-promoting role for B cells in solid tumor development was also revealed in transgenic mice expressing tumor necrosis factor (TNF)-receptor-associated factor 3 (TRAF3) in lymphocytes [88]. TRAF3+ lymphocytes induce humoral immune responses leading to chronic inflammation and a significantly elevated incidence of squamous cell carcinomas [88]. These experimental findings indicate that, in the absence of an initiating oncogenic event, B lymphocyte-mediated chronic inflammation is sufficient to foster solid tumor formation. In contrast, an opposite and anti-tumor immune surveillance role for B lymphocytes has also been demonstrated in a syngeneic melanoma model where deletion of mature B cells by anti-CD20 IgG significantly enhanced tumor growth and metastasis [80], suggesting that the role of B lymphocytes in tumor progression, like CD4+ T lymphocytes, may be context dependent and driven by individual B lymphocyte subtype specificity.
Mechanistically, B cells and humoral immunity can act to modulate solid tumor development by regulation of diverse effector pathways involving secretion of pro-inflammatory, as opposed to regulatory, cytokines, e.g., IL10, TGFβ, inhibition of CTL activity [89], perturbation of TH1/TH2 CD4+ T cell lineages [90, 91], as well as differential recruitment and activation of innate immune cells [89, 92].
Recently, using a transgenic mouse model of inflammation-associated carcinogenesis, i.e., K14-HPV16 mice [93], we revealed a novel pathway by which B lymphocytes enhance squamous carcinogenesis and demonstrated the significance of the B cell/immunoglobulin/FcγR signaling axis. We found that B cells and humoral immunity fostered cancer development by activating Fcγ receptors on resident and recruited myeloid cells [74]. Stromal accumulation of autoantibodies in premalignant skin, through their interaction with activating FcγRs, regulated recruitment, composition, and bioeffector functions of leukocytes, in particular subsets of tumor-promoting polarized myeloid cells in neoplastic tissue which in turn enhanced neoplastic progression and subsequent carcinoma development [74]. A similar pro-tumor role for B cells was recently reported by Ammirante and colleagues who found that B cells are critical for growth of castration-resistant prostate cancer metastasis, not through production of immunoglobulins or regulation of FcγR signaling but instead by delivery of lymphotoxin that in turn activates IKK-α and STAT3 in prostate cancer cells and subsequently stimulates metastasis by an NF-κB-independent, cell-autonomous mechanism [94]. These findings together with other experimental studies support a model in which B cells, through various mechanisms, including humoral immunity, activating FcγRs and IKK, are required for establishing chronic inflammatory programs that promote de novo carcinogenesis.
8 Conclusions
While many factors regulate the propensity of immune cells to promote or repress solid tumor development, polarized adaptive immune responses by B and T lymphocytes can specifically regulate multiple pro-tumor properties of myeloid cells that in turn control many of the “hallmarks” of cancer development [95, 96]. Thus, recognition of the soluble molecules that mediate these important paracrine interactions may represent critical targets to evaluate for anti-cancer therapy. Importantly, targeting of pro-tumor pathways that neutralize M2-type macrophage and/or TH2-type CD4+ T cell responses and therein foster M1 or TH1-type immunity may enhance sensitivity to cytotoxic therapies, including chemo- and radiation therapy, whose durability may be limited by the longevity of the anti-tumor immune responses that they induce.
References
Dunn, G. P., Old, L. J., & Schreiber, R. D. (2004). The immunobiology of cancer immunosurveillance and immunoediting. Immunity, 21(2), 137–148.
Ostrand-Rosenberg, S. (2008). Immune surveillance: a balance between protumor and antitumor immunity. Current Opinion in Genetics & Development, 18(1), 11–18.
Karin, M., Lawrence, T., & Nizet, V. (2006). Innate immunity gone awry: linking microbial infections to chronic inflammation and cancer. Cell, 124(4), 823–835.
de Visser, K. E., Eichten, A., & Coussens, L. M. (2006). Paradoxical roles of the immune system during cancer development. Nature Reviews Cancer, 6, 24–37.
Balkwill, F., Charles, K. A., & Mantovani, A. (2005). Smoldering and polarized inflammation in the initiation and promotion of malignant disease. Cancer Cell, 7(3), 211–217.
Mantovani, A., Allavena, P., Sica, A., & Balkwill, F. (2008). Cancer-related inflammation. Nature, 454(7203), 436–444.
De Palma, M., & Coussens, L. M. (2008). Immune cells and inflammatory mediators as regulators of tumor angiogenesis. In W. D. Figg & J. Folkman (Eds.), Angiogenesis: An integrative approach from science to medicine (pp. 225–238). Springer: New York.
Stout, R. D., & Bottomly, K. (1989). Antigen-specific activation of effector macrophages by IFN-gamma producing (TH1) T cell clones. Failure of IL-4-producing (TH2) T cell clones to activate effector function in macrophages. Journal of Immunology, 142(3), 760–765.
Watkins, S. K., Egilmez, N. K., Suttles, J., & Stout, R. D. (2007). IL-12 rapidly alters the functional profile of tumor-associated and tumor-infiltrating macrophages in vitro and in vivo. Journal of Immunology, 178(3), 1357–1362.
Wakabayashi, O., Yamazaki, K., Oizumi, S., Hommura, F., Kinoshita, I., Ogura, S., et al. (2003). CD4(+) T cells in cancer stroma, not CD8(+) T cells in cancer cell nests, are associated with favorable prognosis in human non-small cell lung cancers. Cancer Science, 94(11), 1003–1009.
Siddiqui, S. A., Frigola, X., Bonne-Annee, S., Mercader, M., Kuntz, S. M., Krambeck, A. E., et al. (2007). Tumor-infiltrating Foxp3-CD4+CD25+ T cells predict poor survival in renal cell carcinoma. Clinical Cancer Research, 13(7), 2075–2081.
DeNardo, D. G., Johansson, M., & Coussens, L. M. (2008). Immune cells as mediators of solid tumor metastasis. Cancer Metastasis Reviews, 27(1), 11–18.
Ruffell, B., Denardo, D. G., Affara, N. I., & Coussens, L. M. (2010). Lymphocytes in cancer development: polarization towards pro-tumor immunity. Cytokine & Growth Factor Reviews, 21, 3–10.
Koebel, C. M., Vermi, W., Swann, J. B., Zerafa, N., Rodig, S. J., Old, L. J., et al. (2007). Adaptive immunity maintains occult cancer in an equilibrium state. Nature, 450(7171), 903–907.
Girardi, M., Glusac, E., Filler, R. B., Roberts, S. J., Propperova, I., Lewis, J., et al. (2003). The distinct contributions of murine T cell receptor (TCR)gammadelta+ and TCRalphabeta+ T cells to different stages of chemically induced skin cancer. Journal of Experimental Medicine, 198(5), 747–755.
Girardi, M., Oppenheim, D., Glusac, E. J., Filler, R., Balmain, A., Tigelaar, R. E., et al. (2004). Characterizing the protective component of the alphabeta T cell response to transplantable squamous cell carcinoma. Journal of Investigative Dermatology, 122(3), 699–706.
Daniel, D., Chiu, C., Giraud, E., Inoue, M., Mizzen, L. A., Chu, N. R., et al. (2005). CD4+ T cell-mediated antigen-specific immunotherapy in a mouse model of cervical cancer. Cancer Research, 65, 2018–2025.
Daniel, D., Meyer-Morse, N., Bergsland, E. K., Dehne, K., Coussens, L. M., & Hanahan, D. (2003). Immune enhancement of skin carcinogenesis by CD4+ T cells. Journal of Experimental Medicine, 197(8), 1017–1028.
Zhou, L., Chong, M. M., & Littman, D. R. (2009). Plasticity of CD4+ T cell lineage differentiation. Immunity, 30(5), 646–655.
Munk, M. E., & Emoto, M. (1995). Functions of T-cell subsets and cytokines in mycobacterial infections. European Respiratory Journal. Supplement, 20, 668s–675s.
Romagnani, S., Parronchi, P., D'Elios, M. M., Romagnani, P., Annunziato, F., Piccinni, M. P., et al. (1997). An update on human Th1 and Th2 cells. International Archives of Allergy and Immunology, 113(1–3), 153–156.
Parker, D. C. (1993). T cell-dependent B cell activation. Annual Review of Immunology, 11, 331–360.
Pollard, J. W. (2004). Tumour-educated macrophages promote tumour progression and metastasis. Nature Reviews. Cancer, 4(1), 71–78.
Aspord, C., Pedroza-Gonzalez, A., Gallegos, M., Tindle, S., Burton, E. C., Su, D., et al. (2007). Breast cancer instructs dendritic cells to prime interleukin 13-secreting CD4+ T cells that facilitate tumor development. Journal of Experimental Medicine, 204(5), 1037–1047.
DeNardo, D. G., Baretto, J. B., Andreu, P., Vasquez, L., Kolhatkar, N., Tawfik, D., et al. (2009). CD4+ T cells regulate pulmonary metastasis of mammary carcinomas by enhancing protumor properties of macrophages. Cancer Cell, 16(2), 91–102.
Chin, Y., Janseens, J., Vandepitte, J., Vandenbrande, J., Opdebeek, L., & Raus, J. (1992). Phenotypic analysis of tumor-infiltrating lymphocytes from human breast cancer. Anticancer Research, 12(5), 1463–1466.
Wilson, C. B., Rowell, E., & Sekimata, M. (2009). Epigenetic control of T-helper-cell differentiation. Nature Reviews. Immunology, 9(2), 91–105.
Weaver, C. T., Harrington, L. E., Mangan, P. R., Gavrieli, M., & Murphy, K. M. (2006). Th17: an effector CD4 T cell lineage with regulatory T cell ties. Immunity, 24(6), 677–688.
Dong, C. (2008). TH17 cells in development: an updated view of their molecular identity and genetic programming. Nature Reviews. Immunology, 8(5), 337–348.
Ouyang, W., Kolls, J. K., & Zheng, Y. (2008). The biological functions of T helper 17 cell effector cytokines in inflammation. Immunity, 28(4), 454–467.
Bettelli, E., Korn, T., Oukka, M., & Kuchroo, V. K. (2008). Induction and effector functions of T(H)17 cells. Nature, 453(7198), 1051–1057.
Wu, S., Rhee, K. J., Albesiano, E., Rabizadeh, S., Wu, X., Yen, H. R., et al. (2009). A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nature Medicine, 15(9), 1016–1022.
Miyahara, Y., Odunsi, K., Chen, W., Peng, G., Matsuzaki, J., & Wang, R. F. (2008). Generation and regulation of human CD4+ IL-17-producing T cells in ovarian cancer. Proceedings of the National Academy of Sciences of the United States of America, 105(40), 15505–15510.
Sfanos, K. S., Bruno, T. C., Maris, C. H., Xu, L., Thoburn, C. J., DeMarzo, A. M., et al. (2008). Phenotypic analysis of prostate-infiltrating lymphocytes reveals TH17 and Treg skewing. Clinical Cancer Research, 14(11), 3254–3261.
Zhang, J. P., Yan, J., Xu, J., Pang, X. H., Chen, M. S., Li, L., et al. (2009). Increased intratumoral IL-17-producing cells correlate with poor survival in hepatocellular carcinoma patients. Journal of Hepatology, 50(5), 980–989.
Numasaki, M., Watanabe, M., Suzuki, T., Takahashi, H., Nakamura, A., McAllister, F., et al. (2005). IL-17 enhances the net angiogenic activity and in vivo growth of human non-small cell lung cancer in SCID mice through promoting CXCR-2-dependent angiogenesis. Journal of Immunology, 175(9), 6177–6189.
Numasaki, M., Fukushi, J., Ono, M., Narula, S. K., Zavodny, P. J., Kudo, T., et al. (2003). Interleukin-17 promotes angiogenesis and tumor growth. Blood, 101(7), 2620–2627.
Martin-Orozco, N., Muranski, P., Chung, Y., Yang, X. O., Yamazaki, T., Lu, S., et al. (2009). T helper 17 cells promote cytotoxic T cell activation in tumor immunity. Immunity, 31(5), 787–798.
Carreras, J., Lopez-Guillermo, A., Fox, B. C., Colomo, L., Martinez, A., Roncador, G., et al. (2006). High numbers of tumor-infiltrating FOXP3-positive regulatory T cells are associated with improved overall survival in follicular lymphoma. Blood, 108(9), 2957–2964.
Hiraoka, N., Onozato, K., Kosuge, T., & Hirohashi, S. (2006). Prevalence of FOXP3+ regulatory T cells increases during the progression of pancreatic ductal adenocarcinoma and its premalignant lesions. Clinical Cancer Research, 12(18), 5423–5434.
Petersen, R. P., Campa, M. J., Sperlazza, J., Conlon, D., Joshi, M. B., Harpole, D. H., Jr., et al. (2006). Tumor infiltrating Foxp3+ regulatory T-cells are associated with recurrence in pathologic stage I NSCLC patients. Cancer, 107(12), 2866–2872.
Merlo, A., Casalini, P., Carcangiu, M. L., Malventano, C., Triulzi, T., Menard, S., et al. (2009). FOXP3 expression and overall survival in breast cancer. Journal of Clinical Oncology, 27(11), 1746–1752.
Trzonkowski, P., Szmit, E., Mysliwska, J., Dobyszuk, A., & Mysliwski, A. (2004). CD4+CD25+ T regulatory cells inhibit cytotoxic activity of T CD8+ and NK lymphocytes in the direct cell-to-cell interaction. Clinical Immunology, 112(3), 258–267.
Strauss, L., Bergmann, C., Szczepanski, M., Gooding, W., Johnson, J. T., & Whiteside, T. L. (2007). A unique subset of CD4+CD25highFoxp3+ T cells secreting interleukin-10 and transforming growth factor-beta1 mediates suppression in the tumor microenvironment. Clinical Cancer Research, 13(15 Pt 1), 4345–4354.
Fehervari, Z., & Sakaguchi, S. (2004). CD4+ Tregs and immune control. Journal of Clinical Investigation, 114(9), 1209–1217.
Feuerer, M., Hill, J. A., Mathis, D., & Benoist, C. (2009). Foxp3+ regulatory T cells: differentiation, specification, subphenotypes. Nature Immunology, 10(7), 689–695.
Cao, X., Cai, S. F., Fehniger, T. A., Song, J., Collins, L. I., Piwnica-Worms, D. R., et al. (2007). Granzyme B and perforin are important for regulatory T cell-mediated suppression of tumor clearance. Immunity, 27(4), 635–646.
Tang, Q., & Bluestone, J. A. (2008). The Foxp3+ regulatory T cell: a jack of all trades, master of regulation. Nature Immunology, 9(3), 239–244.
Okamoto, R., Ueno, M., Yamada, Y., Takahashi, N., Sano, H., Suda, T., et al. (2005). Hematopoietic cells regulate the angiogenic switch during tumorigenesis. Blood, 105(7), 2757–2763.
De Palma, M., Venneri, M. A., Galli, R., Sergi Sergi, L., Politi, L. S., Sampaolesi, M., et al. (2005). Tie2 identifies a hematopoietic lineage of proangiogenic monocytes required for tumor vessel formation and a mesenchymal population of pericyte progenitors. Cancer Cell, 8(3), 211–226.
Coussens, L. M., Raymond, W. W., Bergers, G., Laig-Webster, M., Behrendtsen, O., Werb, Z., et al. (1999). Inflammatory mast cells up-regulate angiogenesis during squamous epithelial carcinogenesis. Genes & Development, 13(11), 1382–1397.
Nozawa, H., Chiu, C., & Hanahan, D. (2006). Infiltrating neutrophils mediate the initial angiogenic switch in a mouse model of multistage carcinogenesis. Proceedings of the National Academy of Sciences of the United States of America, 103(33), 12493–12498.
Takakura, N. (2006). Role of hematopoietic lineage cells as accessory components in blood vessel formation. Cancer Science, 97(7), 568–574.
Zou, W. (2005). Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nature Reviews. Cancer, 5(4), 263–274.
Blankenstein, T. (2005). The role of tumor stroma in the interaction between tumor and immune system. Current Opinion in Immunology, 17(2), 180–186.
Bronte, V., Cingarlini, S., Marigo, I., De Santo, C., Gallina, G., Dolcetti, L., et al. (2006). Leukocyte infiltration in cancer creates an unfavorable environment for antitumor immune responses: a novel target for therapeutic intervention. Immunological Investigations, 35(3–4), 327–357.
Condeelis, J., & Pollard, J. W. (2006). Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell, 124(2), 263–266.
Wyckoff, J., Wang, W., Lin, E. Y., Wang, Y., Pixley, F., Stanley, E. R., et al. (2004). A paracrine loop between tumor cells and macrophages is required for tumor cell migration in mammary tumors. Cancer Research, 64(19), 7022–7029.
Lewis, C. E., & Pollard, J. W. (2006). Distinct role of macrophages in different tumor microenvironments. Cancer Research, 66(2), 605–612.
De Palma, M., & Naldini, L. (2006). Role of haematopoietic cells and endothelial progenitors in tumour angiogenesis. Biochimica et Biophysica Acta, 1766(1), 159–166.
Coukos, G., Benencia, F., Buckanovich, R. J., & Conejo-Garcia, J. R. (2005). The role of dendritic cell precursors in tumour vasculogenesis. British Journal of Cancer, 92(7), 1182–1187.
Yang, L., Debusk, L. M., Fukuda, K., Fingleton, B., Green-Jarvis, B., Shyr, Y., et al. (2004). Expansion of myeloid immune suppressor Gr+CD11b+ cells in tumor-bearing host directly promotes tumor angiogenesis. Cancer Cell, 6(4), 409–421.
Serafini, P., Borrello, I., & Bronte, V. (2006). Myeloid suppressor cells in cancer: recruitment, phenotype, properties, and mechanisms of immune suppression. Seminars in Cancer Biology, 16(1), 53–65.
Kisucka, J., Butterfield, C. E., Duda, D. G., Eichenberger, S. C., Saffaripour, S., Ware, J., et al. (2006). Platelets and platelet adhesion support angiogenesis while preventing excessive hemorrhage. Proceedings of the National Academy of Sciences of the United States of America, 103(4), 855–860.
Puxeddu, I., Alian, A., Piliponsky, A. M., Ribatti, D., Panet, A., & Levi-Schaffer, F. (2005). Human peripheral blood eosinophils induce angiogenesis. International Journal of Biochemistry & Cell Biology, 37(3), 628–636.
Takakura, N., Watanabe, T., Suenobu, S., Yamada, Y., Noda, T., Ito, Y., et al. (2000). A role for hematopoietic stem cells in promoting angiogenesis. Cell, 102(2), 199–209.
Hagemann, T., Lawrence, T., McNeish, I., Charles, K. A., Kulbe, H., Thompson, R. G., et al. (2008). “Re-educating” tumor-associated macrophages by targeting NF-kappaB. Journal of Experimental Medicine, 205(6), 1261–1268.
Qian, B., Deng, Y., Im, J. H., Muschel, R. J., Zou, Y., Li, J., et al. (2009). A distinct macrophage population mediates metastatic breast cancer cell extravasation, establishment and growth. PLoS One, 4(8), e6562.
Corthay, A., Skovseth, D. K., Lundin, K. U., Rosjo, E., Omholt, H., Hofgaard, P. O., et al. (2005). Primary antitumor immune response mediated by CD4+ T cells. Immunity, 22(3), 371–383.
Mantovani, A., Sica, A., & Locati, M. (2007). New vistas on macrophage differentiation and activation. European Journal of Immunology, 37(1), 14–16.
Mosser, D. M., & Edwards, J. P. (2008). Exploring the full spectrum of macrophage activation. Nature Reviews. Immunology, 8(12), 958–969.
Wynn, T. A. (2004). Fibrotic disease and the T(H)1/T(H)2 paradigm. Nature Reviews. Immunology, 4(8), 583–594.
Mantovani, A., Sica, A., Sozzani, S., Allavena, P., Vecchi, A., & Locati, M. (2004). The chemokine system in diverse forms of macrophage activation and polarization. Trends in Immunology, 25(12), 677–686.
Andreu, P., Johansson, M., Affara, N. I., Pucci, F., Tan, T., Junankar, S., et al. (2010). FcRgamma activation regulates inflammation-associated squamous carcinogenesis. Cancer Cell, 17, 121–134.
Kapsenberg, M. L., & Kalinski, P. (1999). The concept of type 1 and type 2 antigen-presenting cells. Immunology Letters, 69(1), 5–6.
Fridlender, Z. G., Sun, J., Kim, S., Kapoor, V., Cheng, G., Ling, L., et al. (2009). Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell, 16(3), 183–194.
Hagemann, T., Wilson, J., Burke, F., Kulbe, H., Li, N. F., Pluddemann, A., et al. (2006). Ovarian cancer cells polarize macrophages toward a tumor-associated phenotype. Journal of Immunology, 176(8), 5023–5032.
Corthay, A. (2007). CD4+ T cells cooperate with macrophages for specific elimination of MHC class II-negative cancer cells. Advances in Experimental Medicine and Biology, 590, 195–208.
Newman, K. C., & Riley, E. M. (2007). Whatever turns you on: accessory-cell-dependent activation of NK cells by pathogens. Nature Reviews. Immunology, 7(4), 279–291.
Szekanecz, Z., & Koch, A. E. (2007). Macrophages and their products in rheumatoid arthritis. Current Opinion in Rheumatology, 19(3), 289–295.
Tiemessen, M. M., Jagger, A. L., Evans, H. G., van Herwijnen, M. J., John, S., & Taams, L. S. (2007). CD4+CD25+Foxp3+ regulatory T cells induce alternative activation of human monocytes/macrophages. Proceedings of the National Academy of Sciences of the United States of America, 104(49), 19446–19451.
Dilillo, D. J., Matsushita, T., & Tedder, T. F. (2010). B10 cells and regulatory B cells balance immune responses during inflammation, autoimmunity, and cancer. Annals of the New York Academy of Sciences, 1183, 38–57.
Mizoguchi, A., & Bhan, A. K. (2006). A case for regulatory B cells. Journal of Immunology, 176(2), 705–710.
Tan, T. T., & Coussens, L. M. (2007). Humoral immunity, inflammation and cancer. Current Opinion in Immunology, 19(2), 209–216.
Inoue, S., Leitner, W. W., Golding, B., & Scott, D. (2006). Inhibitory effects of B cells on antitumor immunity. Cancer Research, 66(15), 7741–7747.
Shah, S., Divekar, A. A., Hilchey, S. P., Cho, H. M., Newman, C. L., Shin, S. U., et al. (2005). Increased rejection of primary tumors in mice lacking B cells: inhibition of anti-tumor CTL and TH1 cytokine responses by B cells. International Journal of Cancer, 117(4), 574–586.
Barbera-Guillem, E., Nelson, M. B., Barr, B., Nyhus, J. K., May, K. F., Jr., Feng, L., et al. (2000). B lymphocyte pathology in human colorectal cancer. Experimental and clinical therapeutic effects of partial B cell depletion. Cancer Immunology and Immunotherapy, 48(10), 541–549.
Zapata, J. M., Llobet, D., Krajewska, M., Lefebvre, S., Kress, C. L., & Reed, J. C. (2009). Lymphocyte-specific TRAF3 transgenic mice have enhanced humoral responses and develop plasmacytosis, autoimmunity, inflammation, and cancer. Blood, 113(19), 4595–4603.
Qin, Z., Richter, G., Schuler, T., Ibe, S., Cao, X., & Blankenstein, T. (1998). B cells inhibit induction of T cell-dependent tumor immunity. Nature Medicine, 4(5), 627–630.
Schreiber, H., Wu, T. H., Nachman, J., & Rowley, D. A. (2000). Immunological enhancement of primary tumor development and its prevention. Seminars in Cancer Biology, 10(5), 351–357.
Wijesuriya, R., Maruo, S., Zou, J. P., Ogawa, M., Umehara, K., Yamashita, M., et al. (1998). B cell-mediated down-regulation of IFN-gamma and IL-12 production induced during anti-tumor immune responses in the tumor-bearing state. International Immunology, 10(8), 1057–1065.
Barbera-Guillem, E., May, K. F., Jr., Nyhus, J. K., & Nelson, M. B. (1999). Promotion of tumor invasion by cooperation of granulocytes and macrophages activated by anti-tumor antibodies. Neoplasia, 1(5), 453–460.
Coussens, L. M., Hanahan, D., & Arbeit, J. M. (1996). Genetic predisposition and parameters of malignant progression in K14- HPV16 transgenic mice. American Journal of Pathology, 149(6), 1899–1917.
Ammirante, M., Luo, J. L., Grivennikov, S., Dedospasov, S., & Karin, M. (2010). B-cell-derived lymphotoxin promotes castration-resistant prostate cancer. Nature, 464, 302–306.
Hanahan, D., & Weinberg, R. A. (2000). The hallmarks of cancer. Cell, 100(1), 57–70.
Colotta, F., Allavena, P., Sica, A., Garlanda, C., & Mantovani, A. (2009). Cancer-related inflammation, the seventh hallmark of cancer: links to genetic instability. Carcinogenesis, 30(7), 1073–1081.
Acknowledgements
The authors acknowledge support from the American Cancer Society (PF-07-264-01 to DD) and the Cancer Research Institute (PA) and grants from the NIH/NCI and Department of Defense (W81XWH-06-1-0416, PR080717) to LMC.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
DeNardo, D.G., Andreu, P. & Coussens, L.M. Interactions between lymphocytes and myeloid cells regulate pro- versus anti-tumor immunity. Cancer Metastasis Rev 29, 309–316 (2010). https://doi.org/10.1007/s10555-010-9223-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10555-010-9223-6