
Abstract
Purpose
We evaluated the safety and efficacy of long-term administration of dexmedetomidine in patients in the intensive care unit (ICU). Primary endpoint was the incidence of hypotension, hypertension, and bradycardia. Secondary endpoints were withdrawal symptoms, rebound effects, the duration of sedation with Richmond Agitation-Sedation Scale (RASS) ≤ 0 relative to the total infusion time of dexmedetomidine, and the dose of additional sedatives or analgesics.
Methods
Dexmedetomidine 0.2–0.7 μg/kg/h was continuously infused for maintaining RASS ≤ 0 in patients requiring sedation in the ICU. Safety and efficacy of short-term (≤24 h) and long-term (>24 h) dexmedetomidine administration were compared.
Results
Seventy-five surgical and medical ICU patients were administered dexmedetomidine. The incidence of hypotension, hypertension, and bradycardia that occurred after 24 h (long-term) was not significantly different from that occurring within 24 h (short-term) (P = 0.546, 0.513, and 0.486, respectively). Regarding withdrawal symptoms, one event each of hypertension and headache occurred after the end of infusion, but both were mild in severity. Increases of mean arterial blood pressure and heart rate after terminating the infusion of dexmedetomidine were not associated with the increasing duration of its infusion. The ratio of duration with RASS ≤ 0 was ≥ 85 % until day 20, except day 9 (70 %) and day 10 (75 %). There was no increase in the dose of additional sedatives or analgesics after the first 24-h treatment period.
Conclusions
Long-term safety of dexmedetomidine compared to its use for 24 h was confirmed. Dexmedetomidine was useful to maintain an adequate sedation level (RASS ≤ 0) during long-term infusion.
Similar content being viewed by others

Introduction
The importance of optimizing the levels of sedation in critical care has been increasingly recognized [1]. Many intensive care experts are focusing on maintaining a targeted “ideal” sedation level according to the individual patient’s condition to avoid adverse events such as prolonged mechanical ventilation, respiratory depression, pneumonia, delirium, psychological problems, and increased treatment costs resulting from oversedation [1–5]. However, optimizing the levels of sedation in intensive care unit (ICU) patients has been challenging, particularly in those requiring long-term sedation, frequently accompanied with severe conditions, and difficult to manage [6]. Although propofol and midazolam have been commonly used for long-time sedation, oversedation and respiratory depression have been regarded as unavoidable complications [4, 5, 7].
Dexmedetomidine is a selective alpha-2-adrenoceptor agonist. It exerts both sedative and analgesic effects via mechanisms different from other sedatives such as midazolam and propofol, and provides sedation characterized by prompt response to stimuli with no respiratory depression [8–11]. Although there have been several reports showing the effects of dexmedetomidine on long-term sedation [12–14], no prospective study has compared the safety and efficacy of short-term (within 24 h) and long-term (longer than 24 h) administration of dexmedetomidine for sedation in the ICU. We performed a prospective, multicenter trial to investigate the safety and efficacy of dexmedetomidine for long-term sedation in surgical and medical ICU patients.
Materials and methods
Study design
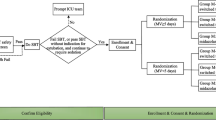
This was a prospective, single-arm, open-label, multicenter, phase III clinical study conducted at ten investigational sites in Japan between October 2007 and June 2008, aimed to obtain an approval for the long-term use (>24 h) of dexmedetomidine in Japan. It was approved by the Institutional Review Board at each site, and all the patients or legally acceptable representatives provided their written informed consent before enrollment. The study was conducted according to the Japanese Pharmaceutical Affairs Law, Japanese Good Clinical Practice, and relevant regulatory standards, and has been registered in ClinicalTrials.gov (NCT00526760) before recruitment of the first subject.
Subjects
Inclusion criteria were patients admitted in either surgical or medical ICU aged ≥20 years, requiring mechanical ventilation and estimated duration of sedation >24 h, with American Society of Anesthesiologists physical status I to III (only those in surgical ICU). Exclusion criteria were patients with serious trauma in the central nervous system, terminal illness with life expectancy ≤30 days, with bleeding probably requiring surgical hemostasis, drug overdose within the last 30 days before study entry, pregnancy/lactation, contraindication to alpha-2-adrenoceptor agonists or antagonists, or difficulty in data collection or completing the study protocol. Patients required neuromuscular blocking agents except for tracheal intubation, received alpha-2-adrenoceptor agonists or antagonists within the last 30 days before participation in the study, patients who had participated in a trial with any experimental drug within 30 days before their admission into the ICU, or patients who had any symptom or factor that might increase the risk to the patients by participating in the study were also excluded.
Treatment
Decision to start and terminate the infusion of dexmedetomidine was made by the investigators or sub-investigators. It was administered at 0.2–0.7 μg/kg/h for maintaining the sedation levels with Richmond Agitation-Sedation Scale (RASS) ≤ 0 [15]. Maximum duration of infusion was 28 days, and restarting infusion after termination was allowed within this limit. It was not necessary to discontinue the administration of the other sedatives or analgesics before starting infusion of dexmedetomidine. If necessary, additional sedatives and analgesics were given after assessing RASS or pain, respectively. Pain was assessed by direct communication with the patients or by an observation of clinical symptoms such as sweating, tachycardia, or hypertension. A 24-h observation period followed the dose administration. The patients were also followed for serious adverse events for 30 days after the end of the infusion.
Efficacy and safety evaluation
The primary endpoint was the incidence rates of treatment-related hypotension, hypertension, and bradycardia, defined in the protocol as (1) hypotension: systolic blood pressure (SBP) < 60 mmHg, diastolic blood pressure (DBP) < 40 mmHg or decrease of SBP by ≥ 50 % from the baseline, requiring infusion or increase of the dose of vasopressors or fluid infusion ≥ 500 ml within 1 h; (2) hypertension: SBP > 180 mmHg, DBP > 100 mmHg, or increase of SBP by ≥ 50 % from the baseline, requiring infusion or increasing the dose of antihypertensive agents; and (3) bradycardia: heart rate (HR) < 40 bpm or decrease by ≥ 50 % from the baseline, requiring infusion or increase the dose of positive chronotropic medications or the use of a pacemaker. Treatment-related adverse events were defined as all the adverse events except those that were deemed “not related” to dexmedetomidine.
Secondary safety assessments included adverse events, withdrawal assessments of the incidence rates of withdrawal symptom-related adverse events (including increased blood pressure, tachycardia, nausea/vomiting, headache, tremor, anxiety, sweating, or agitation), and rebound assessments of the post-infusion changes in mean arterial blood pressure (MBP), HR, and rate-pressure product (RPP). If clinically important abnormal values were observed in hematology and blood chemistry, they were to be reported as adverse events. As with the primary endpoint, treatment-related adverse events were defined as all of the adverse events except that were deemed “not related” to dexmedetomidine.
Secondary efficacy endpoints included the ratio of duration with RASS ≤ 0 to the total duration of infusion of dexmedetomidine, and the dosage of additional sedatives and analgesics. Dexmedetomidine characteristic sedation level corresponds to a RASS of 0 to −2. However, this was a long-term study in ICU patients with a critical condition who would sometimes require deep sedation (RASS < −2), and the target sedation level during the infusion was set as RASS < 0.
Statistical methods
Sample size was determined to detect at least one incidence of treatment-related hypotension, hypertension, or bradycardia. Assuming the incidence of bradycardia to be 5 %, the lowest among those events, 59 patients would be required to detect at least one incidence of 5 % treatment-related adverse events with a 95 % probability. Taking into account 20 % of the dropout cases, 80 patients were estimated as the sample size. It was also planned that approximately 15 % of the medical ICU patients would be enrolled.
For the primary safety analysis, the incidence rates per person per day of treatment-related adverse events including protocol-defined hypotension, hypertension, and bradycardia were calculated by dividing the number of those events by the sum of days of treatment for all patients including the 24-h observation period. The incidence rate during the first 24 h was compared with that after 24 h using the Sumi and Tango method of the score test [16]. In the secondary analysis, the incidence rates of the other treatment-related adverse events were analyzed as described for primary analysis. Descriptive statistics were used in the other assessments. The analysis was on the basis of the full analysis set of patients, which was defined as all the patients who received dexmedetomidine treatment. The level of significance in all statistical analysis was set at α = 0.05 (two-tailed).
Results
Patient demographics
A full analysis set consisted of 75 patients who received dexmedetomidine. Totally, 5 patients dropped out before dexmedetomidine treatment because of change in surgery date, persistent hemorrhage after surgery, or withdrawal of consent, and were excluded from the full analysis set. Of 75 patients, 52 (69.3 %) were surgical ICU and 23 (30.7 %) were medical ICU patients, respectively (Table 1). The medical ICU patients required a longer period of sedation compared to the surgical ICU patients. Maximum duration of dexmedetomidine treatment in the surgical ICU patients and the medical ICU patients was 5.6 and 19.9 days, respectively. Sixty-one of the 75 patients (81.3 %) received dexmedetomidine treatment both before and after extubation. Two of the 52 surgical ICU patients discontinued dexmedetomidine infusion during the first 24 h because of bradycardia or postoperative bleeding (Table 2).
Safety
There were no differences in the incidence rates of treatment-related hypotension, hypertension, or bradycardia defined in the protocol, expressed as per person per day between the first 24 h and after 24 h (Table 3). There were also no differences in those values between the surgical and medical ICU patients.
The total incidence rate of treatment-related adverse events expressed as per person per day that occurred within 24 h was significantly higher than that after 24 h (Table 4). There were no differences in the incidence of each treatment-related adverse event within and after 24 h, with the exception that the incidence of increased blood pressure was higher within 24 h compared with that after 24 h. No treatment-related respiratory depression occurred. Three of 75 patients (4.0 %) developed delirium. One of three events was deemed as probably not related to dexmedetomidine, and the patient recovered 6 days after the onset of symptoms. Another two events were deemed as not related to dexmedetomidine, and the patients recovered about 5 h and 9 days after the onset of symptoms, respectively. Seven patients had died after the end of the dexmedetomidine infusion of respiratory failure, cardiac failure, pneumonia aspiration, multiorgan failure, or sepsis. These events were not considered related to dexmedetomidine infusion, and no other serious adverse events related to dexmedetomidine infusion were observed.
A total of 13 adverse events related to withdrawal symptoms were observed in 9 of 75 patients, and all the adverse events were mild with the exception of 1 moderate headache event (Table 5). One event each of increased blood pressure and headache were considered treatment related, and each event was mild in severity. MBP, HR, and RPP modestly increased after the termination of long-term infusion of dexmedetomidine. Changes were not associated with the increasing duration of dexmedetomidine infusion (Figs. 1, 2, 3).

Mean arterial blood pressure after terminating infusion of dexmedetomidine in patients receiving dexmedetomidine for ≤2 days (n = 38) (a), 3–5 days (n = 24) (b), or >5 days (n = 13) (c). Values are expressed as mean ± SD of 37 or 38 (a), 24 (b), and 12 or 13 (c) individuals

Heart rate after terminating infusion of dexmedetomidine in patients receiving dexmedetomidine for ≤2 days (n = 38) (a), 3–5 days (n = 24) (b), or >5 days (n = 13) (c). Values are expressed as mean ± SD of 37 or 38 (a), 24 (b), and 12 or 13 (c) individuals

Rate–pressure product after terminating infusion of dexmedetomidine in patients receiving dexmedetomidine for ≤2 days (n = 38) (a), 3–5 days (n = 24) (b), or >5 days (n = 13) (c). Values are expressed as mean ± SD of 37 or 38 (a), 24 (b), and 12 or 13 (c) individuals
Efficacy
During administration of the study drug, the patients were within the target sedation range (RASS ≤ 0) 85 % of the time, except on days 9–10. On days 9–10, a medical ICU patient with agitation (including tube pulling and aggressive behavior) and another patient with daytime arousal (RASS > 0) were observed, and the ratio of duration in the target sedation range decreased to approximately 70–75 % (Fig. 4).

The ratio of duration with the Richmond Agitation-Sedation Scale ≤ 0 during dexmedetomidine treatment was calculated for each patient and mean ± SD values were analyzed. There were 75 patients on day 1, 73 on day 2, 37 on day 3, 26 on day 4, 18 on day 5, 13 on day 6, 11 on day 7, 9 on day 8, 8 on days 9–12, 5 on day 13, 4 on days 14–15, 2 on days 16–18, and 1 on days 19–20
The number of patients who required additional sedatives or analgesics did not increase after 24 h compared to the first 24 h (Table 6). Forty of 75 patients (53.3 %) and 24 of 73 patients (32.9 %) required additional sedatives during the first 24 h and after 24 h, respectively, and 21 of 75 patients (28.0 %) and 19 of 73 patients (26.0 %) required additional analgesics during the first 24 h and after 24 h, respectively.
There was no increase in the dose of additional sedatives or analgesics after 24 h administration (Tables 7, 8). Propofol and midazolam were administered to many patients as additional sedatives. Although neither fentanyl nor haloperidol is a sedative, some patients were administered these drugs for sedation. Fentanyl, buprenorphine, pentazocine, or other analgesics were administered for analgesia.
Discussion
The purpose of this study was to evaluate the safety and efficacy of dexmedetomidine for long-term use. We compared the safety and efficacy of dexmedetomidine during the initial period of ≤24 h and the subsequent period. This design and the approach in this prospective study were unique.
In contrast to other sedatives, dexmedetomidine is not associated with respiratory depression [10, 11] and can be administered continuously throughout intubation as well as after extubation. Dexmedetomidine provides a light to moderate level of sedation with the unique feature of arousability [9]. Propofol and midazolam are not typically used after extubation because of the effects of respiratory depression and potential to produce deeper sedation [17, 18]. Therefore, it was decided not to use either as a comparator. Using a placebo as a comparator was denied because of ethical considerations.
Patients who require long-term sedation are typically in more critical condition compared to patients who require short-term sedation, and they sometimes need deep sedation. When deep sedation is required in the usual ICU setting, other sedatives may be used alone or concomitantly with dexmedetomidine. Therefore, the concomitant use of other sedatives as in the usual ICU setting was allowed in this study. In this study, it was considered more important to conduct a long-term investigation according to its use in the usual ICU setting.
Long-term infusion of dexmedetomidine was well tolerated in both surgical and medical ICU patients. The results of this study showed no increase in treatment-related hypotension, hypertension, bradycardia, or other adverse events during a long-term administration period compared to the initial 24 h of treatment. Although MBP, HR, and RPP modestly increased after the termination of dexmedetomidine, the changes were not associated with the increasing duration of dexmedetomidine infusion. There was no evidence suggesting a withdrawal syndrome or rebound effect, which was a concern after the termination of long-term administration of an α2-receptor agonist. Tapering of the dexmedetomidine dose was not necessary, consistent with previous studies [13, 14]. The ratio of duration with RASS ≤ 0 to the total duration of infusion of dexmedetomidine did not decrease after 24 h. Furthermore, neither the number of patients who required additional sedatives/analgesics nor the dose of additional sedatives/analgesics increased over time.
Infusion of a loading dose is required to rapidly increase the plasma concentration of dexmedetomidine; however, it may be accompanied with adverse effects such as hypertension [8, 10]. Although loading infusion was an option for this study and was available at the investigator’s discretion, no patients had received a loading dose. In surgical ICU patients, study drug administration was initiated when the residual effect of anesthesia during surgery was observed. In medical ICU patients, study drug administration was initiated when the effects of other sedatives were still sufficient. While the patients were sedated, the other sedatives were switched to dexmedetomidine or dexmedetomidine were concomitantly administered with the other sedatives. Thus, no loading dose was necessary.
This study included two patients under noninvasive positive pressure ventilation (NPPV) in the medical ICU. Sedation with dexmedetomidine is desirable in these patients as those receiving NPPV should be conscious to minimize the risk of aspiration pneumonia from lack of airway protection [19–21]. On the other hand, these patients experience discomfort and may develop agitation during NPPV from the use of a face mask. In the present study, dexmedetomidine provided adequate sedation in patients receiving NPPV without any evidence of respiratory depression.
Although the post-extubation period was not the main focus in this study and there were no separate sub-analysis data for the post-extubation period only, the efficacy and safety evaluations included not only the intubation period but also the post-extubation period. Of the 75 patients, 61 (81.3 %) received dexmedetomidine after extubation. Long-term use of dexmedetomidine after extubation in these patients was effective, and no adverse event indicating respiratory depression was observed.
There have been several previous reports that dexmedetomidine reduces the incidence of delirium [13, 22]. In this study, 3 of 75 patients (4.0 %) developed delirium, 1 of which events was deemed as probably not related and the others as not related to dexmedetomidine. However, this study was not a comparative study, and there was no use of the Confusion Assessment Method for the ICU [23] to assess delirium. Therefore, we cannot report on the effects of dexmedetomidine on delirium.
Oversedation leads to poor patient prognosis and increased treatment costs [1–5]. Therefore, it is desirable to avoid oversedation and to maintain the patient at an ideal sedation level. Additionally, the ideal level of sedation differs for each patient because the condition of patients managed in ICU settings is highly variable. In this study, investigators used dexmedetomidine as a fundamental sedative to provide a light to moderate level of sedation (in which patients were easily arousable and cooperative). Other sedatives were concomitantly administered, not only when sedation management was difficult with dexmedetomidine alone but also when deep sedation was necessary. As a result, 61.6 % and 38.4 % of patients received additional sedatives and analgesics, respectively.
Although the interaction of concomitant sedatives needs to be carefully monitored, the concomitant use of other sedatives with dexmedetomidine provides benefits in long-term use, as it utilizes each of the sedative’s properties as needed. Other sedatives commonly used for long-term ICU sedation include midazolam and propofol. Midazolam has less vasodilatory effect compared to dexmedetomidine or propofol [24]. However, long-term use of midazolam demonstrates significant interindividual variation in pharmacokinetics and produces an active metabolite, which results in a prolonged recovery to consciousness after long-term treatment [25, 26]. In addition, a patient may also develop tolerance after long-term use of midazolam [24, 27]. Long-term use of propofol has a short elimination half-life and rapid offset to consciousness [25]. However, long-term use of propofol includes an increased risk of infection by the same route, a risk of excessive blood lipids associated with the lipid emulsion formulation, development of tolerance, and propofol infusion syndrome [7, 17, 27, 28]. Potential advantages of dexmedetomidine for long-term use include the arousability feature, and that it is not associated with respiratory depression, both of which can facilitate weaning and extubation. Dexmedetomidine also has the potential to reduce the incidence of delirium, which increases with prolonged ICU stay [13, 22]. The concomitant uses of other sedatives or analgesics were not increased over time, and the majority of patients were maintained at the target sedation levels without any increase in dose, suggesting that there was no development in tolerance. A potential disadvantage of dexmedetomidine is that it should be used very cautiously in patients with hypotension and/or bradycardia [14, 28].
Conclusions
The long-term safety of dexmedetomidine compared to its use for 24 h was confirmed. Dexmedetomidine was useful to maintain adequate sedation levels (RASS ≤ 0) in both surgical and medical ICU patients during long-term infusion. No clinically significant withdrawal symptoms or rebound effects were observed after the end of long-term treatment. The ratio of duration with RASS ≤ 0 did not decrease after the first 24 h administration, and there was no increase in dose of additional sedatives or analgesics, suggesting no tolerance occurred. Considering its unique properties, investigators used dexmedetomidine as the fundamental sedative, and additional sedatives and analgesics were added based on each patient’s condition.
References
Barr J, Fraser GL, Puntillo K, Ely EW, Gélinas C, Dasta JF, Davidson JE, Devlin JW, Kress JP, Joffe AM, Coursin DB, Herr DL, Tung A, Robinson BR, Fontaine DK, Ramsay MA, Riker RR, Sessler CN, Pun B, Skrobik Y, Jaeschke R. American College of Critical Care Medicine. Clinical practice guidelines for the management of pain, agitation, and delirium in adult patients in the intensive care unit. Crit Care Med. 2013;41:263–306.
Rello J, Diaz E, Roque M, Vallés J. Risk factors for developing pneumonia within 48 hours of intubation. Am J Respir Crit Care Med. 1999;159:1742–6.
Jones C, Griffiths RD, Humphris G, Skirrow PM. Memory, delusions, and the development of acute posttraumatic stress disorder-related symptoms after intensive care. Crit Care Med. 2001;29:573–80.
Devlin JW. The pharmacology of oversedation in mechanically ventilated adults. Curr Opin Crit Care. 2008;14:403–7.
Practice guidelines for sedation and analgesia by non-anesthesiologists. Anesthesiology. 2002;96:1004–17.
Arnold HM, Hollands JM, Skrupky LP, Mice ST. Optimizing sustained use of sedation in mechanically ventilated patients: focus on safety. Curr Drug Saf. 2010;5:6–12.
Blanchard AR. Sedation and analgesia in intensive care. Medications attenuate stress response in critical illness. Postgrad Med 2002;111:59–60, 63–64, 67–70.
Kamibayashi T, Maze M. Clinical uses of α2-adrenergic agonists. Anesthesiology. 2000;93:1345–9.
Hall JE, Uhrich TD, Barney JA, Arain SR, Ebert TJ. Sedative, amnestic, and analgesic properties of small-dose dexmedetomidine infusions. Anesth Analg. 2000;90:699–705.
Ebert TJ, Hall JE, Barney JA, Uhrich TD, Colinco MD. The effects of increasing plasma concentrations of dexmedetomidine in humans. Anesthesiology. 2000;93:382–94.
Hsu YW, Cortinez LI, Robertson KM, Keifer JC, Sum-Ping ST, Moretti EW, Young CC, Wright DR, Macleod DB, Somma J. Dexmedetomidine pharmacodynamics. Part I: Crossover comparison of the respiratory effects of dexmedetomidine and remifentanil in healthy volunteers. Anesthesiology. 2004;101:1066–76.
Jakob SM, Ruokonen E, Grounds RM, Sarapohja T, Garratt C, Pocock SJ, Bratty JR, Takala J. Dexmedetomidine vs midazolam or propofol for sedation during prolonged mechanical ventilation: two randomized controlled trials. JAMA. 2012;307:1151–60.
Riker RR, Shehabi Y, Bokesch PM, Ceraso D, Wisemandle W, Koura F, Whitten P, Margolis BD, Byrne DW, Ely EW, Rocha MG. Dexmedetomidine vs. midazolam for sedation of critically ill patients: a randomized trial. JAMA. 2009;301:489–99.
Shehabi Y, Ruettimann U, Adamson H, Innes R, Ickeringill M. Dexmedetomidine infusion for more than 24 hours in critically ill patients: sedative and cardiovascular effects. Intensive Care Med. 2004;30:2188–96.
Sessler CN, Gosnell MS, Grap MJ, Brophy GM, O’Neal PV, Keane KA, Tesoro EP, Elswick RK. The Richmond Agitation-Sedation Scale. Validity and reliability in adult intensive care unit patients. Am J Respir Crit Care Med. 2002;166:1338–44.
Sumi M, Tango T. Inference on the rate ratio of recurrent events for the matched pairs design. Stat Med. 2010;29:3186–93.
Diprivan [Package Insert]. AstraZeneca Pharmaceuticals. 2004.
Midazolam hydrochloride [Package Insert]. Lake Forest, IL: Hospira, Inc. 2005.
Akada S, Takeda S, Yoshida Y, Nakazato K, Mori M, Hongo T, Tanaka K, Sakamoto A. The efficacy of dexmedetomidine in patients with noninvasive ventilation: a preliminary study. Anesth Analg. 2008;107:167–70.
Takasaki Y, Kido T, Semba K. Dexmedetomidine facilitates induction of noninvasive positive pressure ventilation for acute respiratory failure in patients with severe asthma. J Anesth. 2009;23:147–50.
Scala R. Hypercapnic encephalopathy syndrome: a new frontier for non-invasive ventilation? Respir Med. 2011;105:1109–17.
Maldonado JR, Wysong A, van der Starre PJ, Block T, Miller C, Reitz BA. Dexmedetomidine and the reduction of postoperative delirium after cardiac surgery. Psychosomatics. 2009;50:206–17.
Ely EW, Margolin R, Francis J, May L, Truman B, Dittus R, Speroff T, Gautam S, Bernard GR, Inouye SK. Evaluation of delirium in critically ill patients: validation of the Confusion Assessment Method for the Intensive Care Unit (CAM-ICU). Crit Care Med. 2001;29:1370–9.
Shafer A. Complications of sedation with midazolam in the intensive care unit and a comparison with other sedative regimens. Crit Care Med. 1998;26:947–56.
Carrasco G, Molina R, Costa J, Soler JM, Cabré L. Propofol versus midazolam in short-, medium-, and long-term sedation of critically ill patients. Chest. 1993;103:557–64.
Shelly MP, Mendel L, Park GR. Failure of critically ill patients to metabolize midazolam. Anesthesia. 1987;42:619–26.
Zapantis A, Leung S. Tolerance and withdrawal issues with sedation. Crit Care Nurs Clin N Am. 2005;17:211–23.
Riker RR, Fraser GL. Adverse events associated with sedatives, analgesics, and other drugs that provide patient comfort in the intensive care unit. Pharmacotherapy. 2005;25:8S–18S.
Acknowledgments
The study was funded by Hospira Japan Co., Ltd., Osaka, Japan, and Maruishi Pharmaceutical Co., Ltd., Osaka, Japan, both of which manufacture dexmedetomidine. We thank Dr. Ikuto Yoshiya and Dr. Minoru Nomura for their help in designing the study and interpreting the data and Dr. Toshiro Tango for developing the statistical analysis methods.
Conflict of interest
The corresponding author, Misa Kawai, is an employee of Hospira Japan Co., Ltd., and was involved in designing the study, interpreting the data, and preparing and proofing the manuscript. All other authors report no conflict of interests.
Author information
Authors and Affiliations
Corresponding author
Additional information
For the SEDLOT (Safety and Efficacy of Dexmedetomidine in Long-Term Setting) Study Group.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Ozaki, M., Takeda, J., Tanaka, K. et al. Safety and efficacy of dexmedetomidine for long-term sedation in critically ill patients. J Anesth 28, 38–50 (2014). https://doi.org/10.1007/s00540-013-1678-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00540-013-1678-5