
Abstract
The liver has adapted to the inflow of ingested toxins by the evolutionary development of unique regenerative properties and responds to injury or tissue loss by the rapid division of mature cells. Proliferation of the parenchymal cells, i.e. hepatocytes and epithelial cells of the bile duct, is regulated by numerous cytokine/growth-factor-mediated pathways and is synchronised with extracellular matrix degradation and restoration of the vasculature. Resident hepatic stem/progenitor cells have also been identified in small numbers in normal liver and implicated in liver tissue repair. Their putative role in the physiology, pathophysiology and therapy of the liver, however, is not yet precisely known. Hepatic stem/progenitor cells also known as “oval cells” in rodents have been implicated in liver tissue repair, at a time when the capacity for hepatocyte and bile duct replication is exhausted or experimentally inhibited (facultative stem/progenitor cell pool). Although much more has to be learned about the role of stem/progenitor cells in the physiology and pathophysiology of the liver, experimental analysis of the therapeutic value of these cells has been initiated. Transplantation of hepatic stem/progenitor cells or in vivo pharmacological activation of the pool of hepatic stem cells may provide novel modalities for the therapy of liver diseases. In addition, extrahepatic stem cells (e.g. bone marrow cells) are being investigated for their contribution to liver regeneration. Hepatic progenitor cells derived from embryonic stem cells are included in this review, which also discusses future perspectives of stem cell-based therapies for liver diseases.
Similar content being viewed by others


Stem cells in liver regeneration
Physiological homeostasis of the liver
Under physiological conditions, as few as one out of 2000–3000 hepatocytes divide to maintain the physiological liver mass. Liver damage or loss of liver mass can however extensively stimulate the regenerative capacity until the tissue mass has been restored by the proliferation of mature parenchymal liver cells (Fausto et al. 2006). Up to 75% of surgically removed liver mass can be regenerated within 1 week in rodents (Michalopoulos and DeFrances 1997). Accelerated parenchymal regeneration after necrogenic or surgical loss of liver tissue principally originates from the extensive proliferation of mature parenchymal liver cells rather than from liver stem/progenitor cell proliferation. In a young adult rat or mouse, approximately 95% of hepatocytes replicate during the first 3 days after partial hepatectomy. This proportion drops to approximately 80% in senescent rats (Tsanev 1975).
The newborn liver contains only diploid hepatocytes but polyploidisation and binuclearity occurs rapidly after birth. Fractionation of isolated adult rat hepatocytes based on cell density has yielded subpopulations with “small” mononucleated hepatocytes and “large” hepatocytes with higher ploidy. Hepatocytes with higher ploidy have been shown to reside predominantly in the perivenous areas and to contain more DNA and to exhibit greater maturity. The “smaller” mononucleated hepatocytes are located in the periportal areas, contain less DNA and exhibit greater growth factor responsiveness. The gradient of less complex cells with higher proliferation potential (in vitro) in periportal areas and more mature hepatocytes in perivenous areas has been interpreted as evidence for the existence of a physiological stem cell compartment (Sigal et al. 1995). The “streaming liver hypothesis”, which suggests that the liver lobule is organised in a similar way to the intestinal crypt by containing a stem cell pool arising form the periportal area has, however, been disproved by the observation that nearly all hepatocytes proliferate as a response to injury, regardless of location and ploidy.
Although mature hepatocytes and cholangiocytes represent the first and most important resource for tissue repair, experimental data support the hypothesis that the liver also contains or activates a stem cell compartment (Doyle and Ross 2003; Lechner and Habener 2003; Suzuki et al. 2002).
Resident hepatic stem/progenitor cells
Evidence for the existence and activation of a resident hepatic stem/progenitor cells (rHSPC) compartment has been provided from various murine animal models of “oval cell” proliferation (Alison et al. 1997; Fausto 2004; Thorgeirsson 1996). The general principle underlying oval cell activation is based on a combination of liver injury and the inability of hepatocytes to proliferate in response to damage. For example, if a DNA-damaging agent is administered followed by partial hepatectomy, mitosis of hepatocytes is blocked, and oval-shaped cells emerge from the portal zone. These oval cells play a facultative role in liver regeneration, i.e. they contribute to tissue regeneration only in cases in which adult hepatocyte proliferation is inhibited or exhausted (Fausto and Campbell 2003).
A cell compartment that has not as yet been defined gives rise to these rapid proliferating transient cells that subsequently differentiate into mature hepatocytes and bile duct cells. Anatomically, they arise from areas close to the terminal biliary duct (canals of Hering) of the liver, although additional locations have been described. To date, whether oval cells pre-exist in the tissue or develop from other adult cell types (i.e. bile duct cells) after an injury, is unknown. The restricted potential to differentiate into hepatocytes and cholangiocytes qualifies oval cells more as progenitor cells rather than true stem cells. Cell lines with “non-hepatocyte” phenotypes, which can function as lineage-generating precursors, have been derived from livers of rodents exposed to toxic chemicals or carcinogens and from normal liver with no histological evidence of stem/progenitor cell activation (Azuma et al. 2003; Tsao et al. 1984; Tsuchiya et al. 2007).
The dissection and classification of cell populations on the basis of their molecular heterogeneity have become fundamental working tools in cell biology. For example, the differentiation state, lineage specificity or functional status of a given haematopoietic cell population can be precisely determined and communicated based on their antigenic and molecular properties (Mason et al. 2002). In contrast to the haematopoietic cell lineage, the molecular characterisation of the stem cell compartments in solid organs still suffers from the lack of specific markers that unambiguously label all stem cells and only stem cells, enabling their prospective identification, cultivation and propagation. For a long time, this absence of tissue-specific stem cell markers has posed challenges to the identification and isolation of the “true” liver stem cell. A systematic understanding of the stem cell compartment and their respective cell lineages, however, is of fundamental importance for our understanding of physiological liver regeneration and liver tissue repair after injury and disease. A range of markers has been used to characterise the rHSPC compartment including a host of monoclonal antibodies against cytoskeletal proteins and unknown surface antigens. However, few appropriate cell surface markers that permit the fractionation of viable liver cell sub-populations by fluorescence-activated cell sorting exist. Some candidates have been suggested by a recent report on fetal liver epithelial cells (Nierhoff et al. 2005). Another report on adult rat liver cells describes six surface markers unique to adult liver precursor cells, including CD133 (Yovchev et al. 2007). Nevertheless, even the most basic parameters of resident liver stem/progenitor cells, such as their size, precise location within the liver and morphology, are not yet known in vary species.
For this reason, the stem/progenitor compartment of the liver is assumed to consist of cells with various phenotypes and multiple molecular markers. Most of the markers have been identified by immunohistochemistry from rodent models of oval cell activation. In multiple independent studies, these oval cells have been shown to share molecular markers with adult hepatocytes (albumin, cytokeratins 8 and 18), bile duct cells (cytokeratins 7 and 19, OV-6, A6), fetal hepatoblasts (AFP), and haematopoietic stem cells (Thy -1, Sca-1, c-kit). In a recently published mouse model with transgenic expression of the green fluorescence protein (GFP) under transcriptional control of nestin gene promoter and enhancer elements, Gleiberman and co-workers (2005) have identified, in the normal liver, GFP-marked cells at low frequencies with characteristic properties of oval cells, indicating a pre-formed stem/progenitor cell pool in the liver. Additional molecular markers have been identified by genome-wide analysis of mRNA expression in liver tissue and cells derived from rodent injury models with oval cell activation.
Such studies are beginning to elucidate the molecular markers and signalling pathways involved in stem cell/progenitor activation of the liver. One of these pathways involving stromal-derived factor 1 alpha (SDF-1α) has been shown to activate oval cells through SDF-1α/CXCR4 interactions (Hatch et al. 2002). Other molecular pathways involving cytokines have been identified as mitogenic for oval cells. In the CDE diet model of oval cell activation in mice, the over-expression of tumour necrosis factor (TNF), interleukin-6 (IL-6), oncostatin M and others correlates with the rapid phase of oval cell expansion in the liver. The cytokine TWEAK (TNF-related weak inducer of apoptosis) may also specifically activate the rHSPC compartment in mouse liver through binding of the FN-14 receptor, which is expressed on the surface of biliary epithelial cells. Interestingly, unlike TNF or IL-6, TWEAK does not induce the proliferation of the mature hepatocytes and biliary duct epithelial cells (Jakubowski et al. 2005).
Evidence from several laboratories suggests that hepatic stem/progenitor cells express the “side population” (SP)-phenotype similar to multipotent haematopoietic stem cells. The SP-phenotype of haematopoietic stem cells in mice and humans is largely determined by the expression of a protein known as the ABCG2 (BCRP1), viz. ATP-binding cassette transporter (Scharenberg et al. 2002). The SP-cells in liver suspensions are phenotypically heterogeneous with subsets of these cells expressing diverse markers of haematopoietic cell lineages (Uchida et al. 2001). In animal models of hepatic stem/progenitor cell (oval cell) activation, the mRNA of the ABCG2 transporter increases over time in liver tissue indicating an increasing proportion of hepatic stem cells within the non-parenchymal fraction of cells with the SP-phenotype (Shimano et al. 2003). From the data available so far, we can conclude that cells characterised by the SP-phenotype as the common marker account for most of the stem cell activities in their respective organs.
Protocols for the isolation, culture and propagation of resident liver stem/progenitor cells have been established for several decades. Resulting cell lines have drawn attention, both for their therapeutic potential and for their usefulness in exploring the molecular events surrounding liver development and regeneration. Oval cell lines have mostly been derived from liver of rodents exposed to chemicals and carcinogens. Non-hepatocyte epithelial cell lines have also been propagated from normal livers without evidence for oval cell activation. Resulting cell lines have received the generic name “liver epithelial cell lines”. As an example, postnatal mouse liver cells can now be enriched with clonogenic growth potential by using existing cell surface markers (Li et al. 2006). The cells are isolated by single-cell cloning and express liver progenitor cell markers and some antigens shared by bone-marrow-derived haematopoietic stem cells, such as c-Kit and Thy-1. When cultured as aggregates in Matrigel, the mouse liver progenitor cells differentiate into hepatocytes upon treatment with epithelial growth factor (EGF) or differentiate into biliary lineage cells upon treatment with hepatocyte growth factor (HGF). In the presence of dimethylsulphoxide and Matrigel, the cells acquire predominantly bile-lineage phenotypes. Upon transplantation into CCl4-injured liver, engraftment into liver parenchyma and differentiation into hepatocytes have been demonstrated.
Most of the protocols to induce oval cells from murine liver tissue involve treatment of the animals with carcinogens and are thus not appropriate for the isolation of respective human cell populations. Alternatively, Parent and colleagues have established HepaRG cell lines with a dual differentiation potential from a liver associated with chronic hepatitis C infection (Cerec et al. 2007; Parent et al. 2004). One recent study has reported the successful isolation of a stem cell population from a normal adult human liver. Unlike most of the other studies on oval/progenitor cells, these isolated stem cells are closely related to mesenchymal stem cells and do not express haematopoietic markers (Herrera et al. 2006). So far, none of these protocols to isolate human liver progenitor cells would fulfil the requirements for clinical application.
In the human liver, progenitor cell activation has been associated with a variety of liver diseases and the number of progenitor cells has been related to the severity of the disease (Roskams et al. 2003). In parallel to our knowledge of these rodent models, inhibition of the replication of mature hepatocytes is also inhibited in human liver diseases. Recently, hepatocytes have been shown to become senescent, following telomere shortening, in the cirrhotic stage of a wide variety of chronic human liver diseases (Marshall et al. 2005; Wiemann et al. 2002). Probably, this hepatocyte replicative senescence is in part the result of ongoing proliferation during 20–30 years of chronic liver disease. Chronic inflammation and the presence of growth factors and DNA-damaging agents, such as reactive oxygen species and nitrogen species, also play a role. Several chronic human liver diseases display oval cell proliferation, particularly primary biliary cirrhosis (Bisgaard et al. 2002; Tan et al. 2002). Evidence is increasing that “progenitor cell” proliferation is associated with the evolution of hepatocarcinoma and cholangiocarcinoma (Alison and Lovell 2005; Petersen 2001). However, whether oval cells are simply a marker of carcinogenic disease states or whether oval cells are at particular risk of transformation still remains unclear.
Extrahepatic stem cells as a source for liver cells?
In the last few years, many reports have suggested that extrahepatic stem cells participate in liver regeneration and may be useful for treating many diseases (Alison et al. 2000; Herzog et al. 2003; Lagasse et al. 2000; Petersen et al. 1999; Theise et al. 2000a,b). However, subsequent work by several independent groups has clearly shown that hepatocyte replacement levels after bone marrow transplantation are low (<0.01%), unless the bone-marrow-derived hepatocytes have a selective growth advantage (Cantz et al. 2004; Kanazawa and Verma 2003; Wagers et al. 2002). Furthermore, in most of the cases, fusion with host hepatocytes, rather than transdifferentiation of extrahepatic cells, has been described as the underlying principle (Alvarez-Dolado et al. 2003; Quintana-Bustamante et al. 2006; Vassilopoulos et al. 2003; Wang et al. 2003b; Willenbring et al. 2004). Apparently, extrahepatic cells are not directly involved in liver regeneration but may be a suitable tool to correct a metabolic defect by fusion-mediated additive gene transfer. This will be discussed at the end of the second section of this review.
(Stem) Cell-based therapies for liver disorders
Mature (adult) hepatocytes
Despite their restricted proliferation capacity in vitro, adult hepatocytes show a high proliferation capacity in vivo. Experiments with serial murine hepatocyte transplantations in mice deficient in fumaryl-acetoacetate hydrolase (FAH-) have demonstrated more than 69 cell doublings or a 7.3×1020-fold expansion (Overturf et al. 1997). About 30 years ago, the first animal experiments showed isolated hepatocytes suitable for the treatment of liver diseases. For example, the Gunn rat is a model for UDP-glucuronosyl transferase (UGT1A1) deficiency (Crigler-Najjar syndrome type I) leading to high serum bilirubin levels. After transplantation of primary adult hepatocytes into these animals, the bilirubin level is markedly reduced, although not normalised (Matas et al. 1976). Many other animal models for human liver disorders have also been studied for the potential of hepatocyte transplantation, including metabolic disorders such as Wilson’s disease (Long-Evans cinnamon rats; Yoshida et al. 1996), tyrosinemia (FAH- mice; Overturf et al. 1997), hypercholesterolemia (Watanabe rabbit; Wiederkehr et al. 1990), intrahepatic cholestasis mdr3-/- mice (De Vree et al. 1998), and ornithine transcarbamylase (OTC) deficiency (spf-ash mice; Michel et al. 1993).
Meanwhile, a considerable number of patients treated for metabolic disorders has been reported. In 1998, a 10-year-old girl suffering from Crigler-Najjar syndrome type I was treated with an infusion of 7.5×109 hepatocytes through the portal vein for partial correction of the metabolic defect. By 11 months after transplantation, the need for daily phototherapy had decreased from 10–12 h to 6–7 h and lowered the serum bilirubin from 450 μmol/l to 240 μmol/l (Fox et al. 1998). Under immunosuppression with tacrolimus, no signs of rejection of the cells were observed. Despite the possibility of repeated transplantation, a conventional liver transplantation was successfully performed 2.5 years after the cell transplantation. Some other cases have been described in the last few years, including a newborn boy with OTC deficiency (Horslen et al. 2003), a 4-year-old girl with a peroxisomal biogenesis defect (Refsum disease; Sokal et al. 2003) and a 47-year-old woman with glycogen storage disease type 1a (Muraca et al. 2002). A recent review of Anil Dhawan and colleagues summarises the outcome of 20 treated patients and ends with the conclusion that considerable progress has been made in developing the technique but that hepatocyte transplantation is limited by the available supply of liver tissue. Hepatocytes derived from stem cells might provide alternative sources of cells in the future (Dhawan et al. 2006).
Few studies on liver cell therapy for the treatment of acute liver failure in humans have the intention to bridge the gap until patients until receive orthotopic liver transplantations (Bilir et al. 2000; Schneider et al. 2006; Strom et al. 1997). The main challenges for this approach are the number of transplanted cells, the availability of freshly prepared cells or the quality of cryoconserved cells and the need for immunosuppression to prevent the rejection of the transplanted cells. The last-mentioned point may become more important than has been previously considered because liver failure itself gives a high risk for septic complications, which will be aggravated by immunosuppressive drugs.
Fetal hepatic progenitor cells
Recent studies with transplantations of fetal liver cells show a higher proliferation capacity in the host liver than after treatment with adult hepatocytes. In rats, clusters of 500–1000 cells have been observed after fetal liver cell transplantation and partial hepatectomy for stimulation of liver regeneration. This equals an expansion of approximately nine- to ten-fold after 6 months and a repopulation efficacy of 6%–10%. If the rats have been pre-treated with retrorsine, which blocks endogenous hepatocyte proliferation capacity before transplantation, a repopulation of as high as 80% is achieved (Sandhu et al. 2001). After transplantation of mouse fetal liver cells into 14– to 20-day-old uPA-mice, which develop sub-acute liver failure caused by the expression of the uroplasminogen activator (uPA) under the control of the albumin promoter, donor-derived regeneration nodules were also detectable. They show a mature hepatic phenotype as established by gene expression profiling and a functional integration within in the first 4 weeks after transplantation (Cantz et al. 2003). Thus, fetal liver cells may be suitable for overcoming the limitations in engraftment and to allow a functional correction of the disease phenotype, at least in disorders in which a low percentage of cells with a correct genotype are sufficient. Only a few studies have reported, by using human fetal liver cells in the treatment of acute liver failure, modest clinical improvement (Habibullah et al. 1994). Nevertheless, ethical issues to be considered if fetuses from late abortions are necessary, and as for adult hepatocytes, the need for immunosuppressive regimens will cause additional problems.
Bone-marrow-derived cells
Eight years ago, B. Petersen, M. Alison and N. Theise were among the first to describe the contribution of bone-marrow-derived cells to liver regeneration. They analysed sex-mismatched bone marrow and liver transplantations in rats (Petersen et al. 1999), mice (Theise et al. 2000a) and humans (Alison et al. 2000; Theise et al. 2000b) and were able to show Y-chromosome-positive hepatocytes as single cells or small clusters in the recipients.
Since the liver is the site for haematopoiesis at fetal stages, haematopoietic stem cells might have the capability to generate oval cell and parenchymal liver cells also in the adult organ. In adults, the haematopoietic stem cells reside in the bone marrow and have the ability to reconstitute all haematopoietic cell lineages and to rescue lethally irradiated animals from bone marrow failure. Some of the markers of haematopoietic stem cells, for example, c-kit, stem cell factor (SCF) and Thy-1, are also expressed by the putative resident hepatic stem/progenitor cells, which occur in the liver in response to several modes of injury as described above (Fujio et al. 1994; Petersen et al. 1998, 2003). The first reports about the formation of hepatocytes from haematopoietic stem cells in vivo came up in the late nineties and suggested a wider plasticity of HSC than previously known.
The most convincing study was reported by E. Lagasse, who demonstrated that haematopoietic stem cells (c-kit+, lin-, Sca+ and thy-1low) give rise to functional hepatocytes (Lagasse et al. 2000). The presence of large donor-derived regeneration nodules can rescue mice from a lethal disease of hereditary tyrosinemia, and functional assays have been performed to identify and characterise the differentiated cells. However,this particular model provides strong selection pressure on FAH-positive transplanted cells regulated by repeated withdrawal of the compound NTBC (nitro-trifluoro-methylbenzol-cyclohexanedione). Two lessons were to be learned from his experiments. First, a high selection pressure seemed to be necessary for repopulation of a significant part of the liver with bone-marrow-derived cells. Second, only a few cells gave the origin of the small numbered but big sized regeneration nodules. Hence, two years later, a comment of C. Mitchell and N. Fausto summarises the role of bone-marrow-cell-derived liver regeneration as: “rare, but promising” (Mitchell and Fausto 2002). So far, bone-marrow-derived cells have been used only in non-controlled, non-randomised phase-I clinical trials with a few patients suffering from chronic liver diseases (Gordon et al. 2006; Terai et al. 2006). The authors of these studies report an improvement of liver function in a subset of patients, but these findings should be interpreted carefully, as no control group was investigated. Autologous CD133-positive bone marrow cells have been used to improve the expansion of the residual liver lobe after portal vein embolisation of tumor-bearing liver segments (am Esch et al. 2005; Furst et al. 2007). Compared with a control group, the increase in the liver volume is significantly higher in the CD133-treated group but whether this effect is attributable to cell engraftment and transdifferentiation, to regenerative factors secreted by the transplanted cells or to the method of cell preparation remains unclear.
Therefore, we speculate that bone marrow transplantation without expansion of the resulting donor hepatocytes might not be therapeutically effective and that sophisticated methods have to be applied to increase the number of stem-cell-derived hepatocytes in the recipient liver. Three main determinants could influence the role of stem cells in liver repopulation: (1) the time course, severity and type of liver tissue injury, (2) the expression of homing factors for stem cells in the recipient liver and (3) a sufficient number of circulating haematopoietic stem cells at the time of tissue regeneration. Cantz et al. (2004) have tested two of the three variables by application of the liver toxin carbon tetrachloride (CCl4) to induce liver tissue injury and by mobilising the bone marrow stem cells into the peripheral blood by granulocyte colony-stimulating factor (G-CSF). The authors have found no evidence of an effect of CCl4 and/or stem cell mobilisation regimens on the usual low transdifferentiation rate of bone marrow cells into hepatocytes (Cantz et al. 2004). In another study, Kanazawa and Verma (2003) have addressed the effect of acute and chronic liver injury on bone-marrow-derived hepatocyte formation. They have used three different models of liver injury: (1) CCl4 application to induce toxic liver damage, (2) the albumin-urokinase transgenic mouse model with sub-acute intrinsic liver failure and (3) the hepatitis B transgenic mouse representing a chronic liver disease model. Bone-marrow-derived cells from GFP- or β-galactosidase transgenic mice were transplanted into irradiated sex-mismatched mice. Even 47 weeks after transplantation, only a low number of transgene-marked bone-marrow-derived hepatocytes was detected in any of the recipient mice (Kanazawa and Verma 2003).
In the meantime, several groups have made progress in elucidating the mechanism of the observed stem cell plasticity, which was first considered to be transdifferentiation. Several studies have clearly shown that the correction of the FAH-/- phenotype is attributable to the fusion of bone-marrow-derived cells with host hepatocytes (Alvarez-Dolado et al. 2003; Vassilopoulos et al. 2003; Wang et al. 2003b). In the most detailed study (Willenbring et al. 2004), several controversial points have been addressed: (1) no fusion of mature hepatocytes has been observed, which strongly disproves a transdifferentiation-followed-by-fusion theory, (2) bone marrow reconstitution is not necessary to induce fusion of host hepatocytes and granulocyte-macrophage progenitors, (3) even after intrasplenic transplantation of in vitro differentiated mature macrophages, fusion between donor and host cells is observed. The authors conclude that the administration of fusogenic macrophages to the damaged organ represents a targeted therapeutic strategy (Willenbring et al. 2004). Quintana-Bustamante and co-workers (2006) have applied CCl4 over 3 months followed by injection of pegylated G-CSF for up to 3 weeks in mice that had been irradiated and reconstituted with GFP-marked bone marrow. The authors have demonstrated that the observed bone-marrow-derived hepatocytes do not originate from the differentiation of haematopoietic stem cells but are generated by fusion. The number of bone-marrow-derived hepatocytes is much lower than previously reported and only marginally increased by chronic liver injury and mobilisation of bone marrow stem cells (Quintana-Bustamante et al. 2006).
Thorgeirsson and Grisham (2006) have recently reviewed 77 published reports that examine the ability of haematopoietic cells to generate hepatocytes in the liver. They conclude that haematopoietic cells do not trans-differentiate into hepatocytes and contribute little to hepatocyte formation under physiological or pathological conditions. Although fusion clearly is the only established way to generate hepatocytes from blood stem cells so far, the debate concerning whether this is the only mechanism has not yet been decided (Thorgeirsson and Grisham 2006).
In contradiction to these findings, other groups report bone-marrow-derived liver cells without fusion of haematopoietic donor and host liver cells. In a sophisticated study with the Cre/lox recombinase system, Harris et al. (2004) have shown that bone-marrow-derived epithelial cells occur in the liver, lung, and skin without cell fusion. Whether these cells develop by the transdifferentiation of haematopoietic stem cells or by the differentiation of multipotent epithelial precursors existing in the bone marrow preparation remains to be investigated. The derivation of multipotent adult progenitor cells during the culture of bone marrow cells has been well examined by the group of C. Verfaillie (Reyes et al. 2001) and the feasibility of in vivo (Schwartz et al. 2002) and in vitro (Jiang et al. 2002) differentiation into hepatocytes has been demonstrated. However, the data remain preliminary and functional analyses in terms of transplantation experiments have not yet been performed.
Another group has studied the conversion of haematopoietic stem cells into liver cells in vitro by using a co-culture system in which the stem cells are separated from a CCl4-damaged liver section (Jang et al. 2004). The authors again conclude that microenvironmental cues rather than fusion are responsible for the germ layer switching of the haematopoietic stem cells. Moreover, highly enriched male haematopoietic stem cells were injected, via the tail vein, into irradiated female mice in vivo. A subgroup of mice received CCl4 additionally to increase liver injury. Confocal serial imaging showed mostly diploid (XY) or tetraploid (XXYY) male cells expressing hepatic marker proteins such as albumin and only a few cells with a fusion-related karyotype (XXXY). Aurich et al. (2007) have reported that human bone-marrow-derived mesenchymal stem cells can be pre-differentiated towards a hepatic phenotype in vitro, before transplantation into RAG2-mice. The authors have demonstrated the presence of single human hepatocyte-like cells with no signs of fusion in this experimental setting (Aurich et al. 2007).
Different explanations can be presented for these contradictory results. First, the chronic damage in the FAH-/- mice causes chromosomal instability (Jorquera and Tanguay 2001) that may facilitate fusion events. Second, the type and extent of liver damage (metabolic disorders, irradiation, toxic injury, ischaemia) may play a role. Third, it may be important to transplant very early haematopoietic progenitors in a sufficient number to see conversion into epithelial cells.
These hypotheses may also explain the failure of other groups to show a significant impact of bone-marrow-derived cells on liver regeneration in mice (Cantz et al. 2004; Kanazawa and Verma 2003; Wagers et al. 2002). Although the age of donor mice, the severity of liver damage between different mouse strains (Shi et al. 1997) and the experimental settings are not similar in these published experiments, they clearly show that the use of bone-marrow-derived cells for the treatment of liver diseases is far from clinical application and that additional basic research on reprogramming is needed.
Human umbilical cord blood
Some researchers have argued that stem cells derived from bone marrow or cord blood of human origin exhibit higher plasticity than the respective mouse or rat cells (Di Campli et al. 2004; Ishikawa et al. 2003; Kakinuma et al. 2003; Newsome et al. 2003; Tanabe et al. 2004; Turrini et al. 2005; Wang et al. 2003a). Indeed, several groups have detected high rates of human hepatocyte formation without signs of fusion in xenogenic murine transplantation models. In 2003, Newsome and his colleagues demonstrated that human umbilical cord-blood (hUCB)-derived cells could differentiate into hepatocytes after transplantation into immunodeficient mice (Newsome et al. 2003). In their study, unsorted mononuclear hUCB cells were transplanted into sub-lethally irradiated NOD/SCID mice and the livers were analysed after 1, 4, 6 and 16 weeks for the presence of human hepatocytes. The percentage of human compared with mouse hepatocytes reached an average of 0.011% after 16 weeks, whereas no additional damage to the host liver was inflicted, other than the sub-lethal irradiation before reconstitution of the bone marrow. The absence of mouse and human DNA together within single cells ruled out fusion as the underlying mechanism for the stem cell plasticity and once again favoured the transdifferentiation theory. From these experiments, the questions arose as to whether the hUCB had an even higher plasticity than bone-marrow-derived stem cells or whether the immunodeficient NOD/SCID mice facilitated the transdifferentiation of transplanted stem cells. To investigate these points, mononuclear bone marrow cells from enhanced-GFP transgenic mice and hUCB cells were transplanted, with the same protocol, into NOD/SCID mice and, in addition, hepatocellular damage with CCl4 was administered (Sharma et al. 2005). No donor-derived cells were detectable after transplantation of murine bone marrow cells but some single hepatocytes and even a few clusters of human origin were detected in the hUCB-transplanted mice. However, marker expression was incomplete in these cells. Serial sections of the recipient livers revealed a mixed expression of mouse and human antigens indicating fused “chimerical” mouse/human hepatocytes in the recipient mouse livers (Sharma et al. 2005).
In 2004, human cord-blood-derived multipotent cells were described by Kögler and colleagues( 2004). These “unrestricted somatic stem cells” (USSC) could differentiate into hepatocytes after transplantation into a pre-immune fetal sheep model (Kögler et al. 2004). Subsequently, the fusion of USSC with host hepatocytes was ruled out by using species-specific polymerase chain reaction on micro-dissected tissue samples (G. Kögler, personal communication).
The infusion of particular subsets of haematopoietic stem cells (Liv8-) may be of therapeutic benefit, even in the absence of hepatocyte formation. According to one recently published study, haematopoietic stem cells can partially revert CCl4-induced fibrosis of the mouse liver through the activation of the matrix metalloproteinase-9 and improves the survival of the animals (Sakaida et al. 2004).
Embryonic stem cells
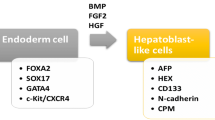
Embryonic stem (ES) cells can be maintained in a state of pluripotency for long periods of time and can be grown in large numbers (Boiani and Scholer 2005; Keller 1995; Rathjen and Rathjen 2001). Under appropriate culture conditions, ES cells spontaneously form all somatic cell types of the adult body, including endoderm-derived cells and hepatic progenitors. The non-directed differentiation of ES cells can be achieved through the formation of embryoid bodies (EB), which contain cells representing lineages derived from the three germ layers: ectoderm, mesoderm and endoderm. Subsequent inoculation of the EB-derived cells onto adherent matrices facilitates differentiation into hepatocyte-like cells in the presence of a variety of growth factors, cytokines and hormones, such as hepatocyte growth factor (HGF), fibroblast growth factor (FGF) and dexamethasone. Significant progress has been made in identifying factors and developing protocols to enforce the differentiation of ES cells into cells of the hepatocyte lineage (Chinzei et al. 2002; Choi et al. 2002; Gouon-Evans et al. 2006; Hamazaki et al. 2001; Heo et al. 2006; Ishizaka et al. 2002; Jones et al. 2002; Kania et al. 2004; Miyashita et al. 2002; Teratani et al. 2005; Yamada et al. 2002; Yin et al. 2002). Hamazaki and co-workers (2001) were among the first to generate a high percentage of hepatocyte precursors from murine ES cell cultures in the presence of growth factors. Miyashita et al. (2002) and Kania et al. (2004) have subsequently published differentiation protocols that direct the maturation of the primitive ES cells towards cells with a distinct and mature hepatic phenotype and that result in a high percentage of ES hepatic progenitor cells (ES-HPC) in culture. Bone morphogenetic protein-4 has recently been identified as mediating an important signal in the development of cells with a hepatic phenotype from ES cells (Gouon-Evans et al. 2006).
The transplantation of contaminating pluripotent ES cells carries the risk of teratoma formation and may mask or inhibit the formation of hepatocytes and normal liver tissue from ES-HPC. Cell-sorting techniques utilising either antibodies directed against lineage-specific surface markers of the desired cell population or cell-type-specific expression of reporter genes have been established to eliminate contaminating undifferentiated ES cells and thus to reduce the risk of teratoma formation (Klug et al. 1996). Another strategy to prevent teratoma formation is to implant the ES-HPC into devices for bio-artificial liver support (Soto-Gutierrez et al. 2006).
Hepatocyte formation derived from sorted ES-HPC has recently been demonstrated in fumaryl-acetoacetate-deficient (FAH-/-) and uroplasminogen-activator transgenic MUP-uPA/SCID mice, clearly indicating the potential of ES-HPC for cellular therapies (Gouon-Evans et al. 2006; Heo et al. 2006). The rate of hepatocyte and liver tissue formation, however, is much lower than that previously reported for transplanted primary adult hepatocytes or fetal hepatoblasts (Fig. 1). The reduced efficacy of hepatocyte and liver tissue formation may have been attributable to lower primary engraftment rates of the ES-HPC into the recipient liver, incomplete differentiation or a combination of both.

Transplantation of ES-derived hepatic progenitor cells. Embryonic stem cells can be differentiated towards a hepatic progenitor cell-phenotype (ES-HPC). After transplantation of wild-type ES-HPCs into immunosuppressed FAH-/- mice, engraftment of single cells (arrowheads) and formation of regeneration nodules after ES-HPC proliferation (arrows) can be observed. The 3-amino-9-ethylcarbazole-substrate staining of the cells demonstrates the presence of FAH-enzyme activity in the FAH-/- recipient liver. Bar 25 μm
Future directions
The transplantation of mature hepatocytes into humans over the past 20 years has provided insights regarding the way in which to treat human liver disease by cellular therapies. The shortage of adult hepatocytes from donated organs has given a strong impetus to the search for alternative cellular resources. Stem-cell-based therapies for the liver, however, have not yet reached the stage of clinical application. Progenitor cell lines derived from normal human liver tissue may be an alternative source of hepatocytes for therapy in the future, although isolation, cultivation and propagation protocols will have to be improved substantially. Human fetal liver progenitor cells have been shown to engraft and proliferate in rodents and could eventually become an important source for liver cell therapy, if fetal liver cells can be successfully expanded in vitro. Currently, transplantation into FAH-/- mice is the only model in which haematopoietic cells have contributed to liver repopulation to a degree relevant for clinical application. The underlying mechanism is fusion with host hepatocytes, providing an additive gene transfer, which rescues the metabolic defect. However, the frequency of in vivo fusion of macrophages and endogenous hepatocytes is below the requirements of most clinical applications, which lack a selection pressure as strong as that in the FAH-/- model.
The application of stem cells in liver therapies seems to be a promising approach for the treatment of liver diseases. However, several issues still have to be addressed to fulfil this promise. We need to identify, both inside and outside of the liver, the stem cell candidates that are able to form hepatocytes in vitro and functional liver tissue after transplantation in vivo. The fundamental molecular pathways involved in the differentiation of hepatocytes and cholangiocytes from stem/progenitor cells, the factors that are responsible for in vitro differentiation of various stem cells into hepatocytes, the mechanisms involved in the fusion of macrophages and hepatocytes and the aspects that can potentially enhance these mechanisms need to be studied in more detail. Finally, the role of hepatic stem/progenitor cells in response to liver injury and in the normal regeneration of liver must be elucidated further. With future progress in research into stem cell biology, hepatic stem/progenitor cells, embryonic and adult extrahepatic stem cells should provide great potential for the treatment of liver disorders.
References
Alison M, Golding M, Lalani EN, Nagy P, Thorgeirsson S, Sarraf C (1997) Wholesale hepatocytic differentiation in the rat from ductular oval cells, the progeny of biliary stem cells. J Hepatol 26:343–352
Alison MR, Lovell MJ (2005) Liver cancer: the role of stem cells. Cell Prolif 38:407–421
Alison MR, Poulsom R, Jeffery R, Dhillon AP, Quaglia A, Jacob J, Novelli M, Prentice G, Williamson J, Wright NA (2000) Hepatocytes from non-hepatic adult stem cells. Nature 406:257
Alvarez-Dolado M, Pardal R, Garcia-Verdugo JM, Fike JR, Lee HO, Pfeffer K, Lois C, Morrison SJ, Alvarez-Buylla A (2003) Fusion of bone-marrow-derived cells with Purkinje neurons, cardiomyocytes and hepatocytes. Nature 425:968–973
Aurich I, Mueller LP, Aurich H, Luetzkendorf J, Tisljar K, Dollinger MM, Schormann W, Walldorf J, Hengstler JG, Fleig WE, Christ B (2007) Functional integration of hepatocytes derived from human mesenchymal stem cells into mouse livers. Gut 56:405–415
Azuma H, Hirose T, Fujii H, Oe S, Yasuchika K, Fujikawa T, Yamaoka Y (2003) Enrichment of hepatic progenitor cells from adult mouse liver. Hepatology 37:1385–1394
Bilir BM, Guinette D, Karrer F, Kumpe DA, Krysl J, Stephens J, McGavran L, Ostrowska A, Durham J (2000) Hepatocyte transplantation in acute liver failure. Liver Transpl 6:32–40
Bisgaard HC, Holmskov U, Santoni-Rugiu E, Nagy P, Nielsen O, Ott P, Hage E, Dalhoff K, Rasmussen LJ, Tygstrup N (2002) Heterogeneity of ductular reactions in adult rat and human liver revealed by novel expression of deleted in malignant brain tumor 1. Am J Pathol 161:1187–1198
Boiani M, Scholer HR (2005) Regulatory networks in embryo-derived pluripotent stem cells. Nat Rev Mol Cell Biol 6:872–884
Cantz T, Zuckerman DM, Burda MR, Dandri M, Goricke B, Thalhammer S, Heckl WM, Manns MP, Petersen J, Ott M (2003) Quantitative gene expression analysis reveals transition of fetal liver progenitor cells to mature hepatocytes after transplantation in uPA/RAG-2 mice. Am J Pathol 162:37–45
Cantz T, Sharma AD, Jochheim-Richter A, Arseniev L, Klein C, Manns MP, Ott M (2004) Reevaluation of bone marrow-derived cells as a source for hepatocyte regeneration. Cell Transplant 13:659–666
Cerec V, Glaise D, Garnier D, Morosan S, Turlin B, Drenou B, Gripon P, Kremsdorf D, Guguen-Guillouzo C, Corlu A (2007) Transdifferentiation of hepatocyte-like cells from the human hepatoma HepaRG cell line through bipotent progenitor. Hepatology 45:957–967
Chinzei R, Tanaka Y, Shimizu-Saito K, Hara Y, Kakinuma S, Watanabe M, Teramoto K, Arii S, Takase K, Sato C, Terada N, Teraoka H (2002) Embryoid-body cells derived from a mouse embryonic stem cell line show differentiation into functional hepatocytes. Hepatology 36:22–29
Choi D, Oh HJ, Chang UJ, Koo SK, Jiang JX, Hwang SY, Lee JD, Yeoh GC, Shin HS, Lee JS, Oh B (2002) In vivo differentiation of mouse embryonic stem cells into hepatocytes. Cell Transplant 11:359–368
Dhawan A, Mitry RR, Hughes RD (2006) Hepatocyte transplantation for liver-based metabolic disorders. J Inherit Metab Dis 29:431–435
Di Campli C, Piscaglia AC, Pierelli L, Rutella S, Bonanno G, Alison MR, Mariotti A, Vecchio FM, Nestola M, Monego G, Michetti F, Mancuso S, Pola P, Leone G, Gasbarrini G, Gasbarrini A (2004) A human umbilical cord stem cell rescue therapy in a murine model of toxic liver injury. Dig Liver Dis 36:603–613
Doyle LA, Ross DD (2003) Multidrug resistance mediated by the breast cancer resistance protein BCRP (ABCG2). Oncogene 22:7340–7358
Esch JS 2nd, Knoefel WT, Klein M, Ghodsizad A, Fuerst G, Poll LW, Piechaczek C, Burchardt ER, Feifel N, Stoldt V, Stockschlader M, Stoecklein N, Tustas RY, Eisenberger CF, Peiper M, Haussinger D, Hosch SB (2005) Portal application of autologous CD133+ bone marrow cells to the liver: a novel concept to support hepatic regeneration. Stem Cells 23:463–470
Fausto N (2004) Liver regeneration and repair: hepatocytes, progenitor cells, and stem cells. Hepatology 39:1477–1487
Fausto N, Campbell JS (2003) The role of hepatocytes and oval cells in liver regeneration and repopulation. Mech Dev 120:117–130
Fausto N, Campbell JS, Riehle KJ (2006) Liver regeneration. Hepatology 43:S45–S53
Fox IJ, Chowdhury JR, Kaufman SS, Goertzen TC, Chowdhury NR, Warkentin PI, Dorko K, Sauter BV, Strom SC (1998) Treatment of the Crigler-Najjar syndrome type I with hepatocyte transplantation. N Engl J Med 338:1422–1426
Fujio K, Evarts RP, Hu Z, Marsden ER, Thorgeirsson SS (1994) Expression of stem cell factor and its receptor, c-kit, during liver regeneration from putative stem cells in adult rat. Lab Invest 70:511–516
Furst G, Esch JS am, Poll LW, Hosch SB, Fritz LB, Klein M, Godehardt E, Krieg A, Wecker B, Stoldt V, Stockschlader M, Eisenberger CF, Modder U, Knoefel WT (2007) Portal vein embolization and autologous CD133+ bone marrow stem cells for liver regeneration: initial experience. Radiology 243:171–179
Gleiberman AS, Encinas JM, Mignone JL, Michurina T, Rosenfeld MG, Enikolopov G (2005) Expression of nestin-green fluorescent protein transgene marks oval cells in the adult liver. Dev Dyn 234:413–421
Gordon MY, Levicar N, Pai M, Bachellier P, Dimarakis I, Al-Allaf F, M’Hamdi H, Thalji T, Welsh JP, Marley SB, Davies J, Dazzi F, Marelli-Berg F, Tait P, Playford R, Jiao L, Jensen S, Nicholls JP, Ayav A, Nohandani M, Farzaneh F, Gaken J, Dodge R, Alison M, Apperley JF, Lechler R, Habib NA (2006) Characterization and clinical application of human CD34+ stem/progenitor cell populations mobilized into the blood by granulocyte colony-stimulating factor. Stem Cells 24:1822–1830
Gouon-Evans V, Boussemart L, Gadue P, Nierhoff D, Koehler CI, Kubo A, Shafritz DA, Keller G (2006) BMP-4 is required for hepatic specification of mouse embryonic stem cell-derived definitive endoderm. Nat Biotechnol 24:1402–1411
Habibullah CM, Syed IH, Qamar A, Taher-Uz Z (1994) Human fetal hepatocyte transplantation in patients with fulminant hepatic failure. Transplantation 58:951–952
Hamazaki T, Iiboshi Y, Oka M, Papst PJ, Meacham AM, Zon LI, Terada N (2001) Hepatic maturation in differentiating embryonic stem cells in vitro. FEBS Lett 497:15–19
Harris RG, Herzog EL, Bruscia EM, Grove JE, Van Arnam JS, Krause DS (2004) Lack of a fusion requirement for development of bone marrow-derived epithelia. Science 305:90–93
Hatch HM, Zheng D, Jorgensen ML, Petersen BE (2002) SDF-1alpha/CXCR4: a mechanism for hepatic oval cell activation and bone marrow stem cell recruitment to the injured liver of rats. Cloning Stem Cells 4:339–351
Heo J, Factor VM, Uren T, Takahama Y, Lee JS, Major M, Feinstone SM, Thorgeirsson SS (2006) Hepatic precursors derived from murine embryonic stem cells contribute to regeneration of injured liver. Hepatology 44:1478–1486
Herrera MB, Bruno S, Buttiglieri S, Tetta C, Gatti S, Deregibus MC, Bussolati B, Camussi G (2006) Isolation and characterization of a stem cell population from adult human liver. Stem Cells 24:2840–2850
Herzog EL, Chai L, Krause DS (2003) Plasticity of marrow-derived stem cells. Blood 102:3483–3493
Horslen SP, McCowan TC, Goertzen TC, Warkentin PI, Cai HB, Strom SC, Fox IJ (2003) Isolated hepatocyte transplantation in an infant with a severe urea cycle disorder. Pediatrics 111:1262–1267
Ishikawa F, Drake CJ, Yang S, Fleming P, Minamiguchi H, Visconti RP, Crosby CV, Argraves WS, Harada M, Key LL Jr, Livingston AG, Wingard JR, Ogawa M (2003) Transplanted human cord blood cells give rise to hepatocytes in engrafted mice. Ann N Y Acad Sci 996:174–185
Ishizaka S, Shiroi A, Kanda S, Yoshikawa M, Tsujinoue H, Kuriyama S, Hasuma T, Nakatani K, Takahashi K (2002) Development of hepatocytes from ES cells after transfection with the HNF-3beta gene. FASEB J 16:1444–1446
Jakubowski A, Ambrose C, Parr M, Lincecum JM, Wang MZ, Zheng TS, Browning B, Michaelson JS, Baetscher M, Wang B, Bissell DM, Burkly LC (2005) TWEAK induces liver progenitor cell proliferation. J Clin Invest 115:2330–2340
Jang YY, Collector MI, Baylin SB, Diehl AM, Sharkis SJ (2004) Hematopoietic stem cells convert into liver cells within days without fusion. Nat Cell Biol 6:532–539
Jiang Y, Jahagirdar BN, Reinhardt RL, Schwartz RE, Keene CD, Ortiz-Gonzalez XR, Reyes M, Lenvik T, Lund T, Blackstad M, Du J, Aldrich S, Lisberg A, Low WC, Largaespada DA, Verfaillie CM (2002) Pluripotency of mesenchymal stem cells derived from adult marrow. Nature 418:41–49
Jones EA, Tosh D, Wilson DI, Lindsay S, Forrester LM (2002) Hepatic differentiation of murine embryonic stem cells. Exp Cell Res 272:15–22
Jorquera R, Tanguay RM (2001) Fumarylacetoacetate, the metabolite accumulating in hereditary tyrosinemia, activates the ERK pathway and induces mitotic abnormalities and genomic instability. Hum Mol Genet 10:1741–1752
Kakinuma S, Tanaka Y, Chinzei R, Watanabe M, Shimizu-Saito K, Hara Y, Teramoto K, Arii S, Sato C, Takase K, Yasumizu T, Teraoka H (2003) Human umbilical cord blood as a source of transplantable hepatic progenitor cells. Stem Cells 21:217–227
Kanazawa Y, Verma IM (2003) Little evidence of bone marrow-derived hepatocytes in the replacement of injured liver. Proc Natl Acad Sci USA 100 (Suppl 1):11850–11853
Kania G, Blyszczuk P, Jochheim A, Ott M, Wobus AM (2004) Generation of glycogen- and albumin-producing hepatocyte-like cells from embryonic stem cells. Biol Chem 385:943–953
Keller GM (1995) In vitro differentiation of embryonic stem cells. Curr Opin Cell Biol 7:862–869
Klug MG, Soonpaa MH, Koh GY, Field LJ (1996) Genetically selected cardiomyocytes from differentiating embronic stem cells form stable intracardiac grafts. J Clin Invest 98:216–224
Kögler G, Sensken S, Airey JA, Trapp T, Muschen M, Feldhahn N, Liedtke S, Sorg RV, Fischer J, Rosenbaum C, Greschat S, Knipper A, Bender J, Degistirici O, Gao J, Caplan AI, Colletti EJ, Almeida-Porada G, Muller HW, Zanjani E, Wernet P (2004) A new human somatic stem cell from placental cord blood with intrinsic pluripotent differentiation potential. J Exp Med 200:123–135
Lagasse E, Connors H, Al-Dhalimy M, Reitsma M, Dohse M, Osborne L, Wang X, Finegold M, Weissman IL, Grompe M (2000) Purified hematopoietic stem cells can differentiate into hepatocytes in vivo. Nat Med 6:1229–1234
Lechner A, Habener JF (2003) Stem/progenitor cells derived from adult tissues: potential for the treatment of diabetes mellitus. Am J Physiol Endocrinol Metab 284:E259–E266
Li WL, Su J, Yao YC, Tao XR, Yan YB, Yu HY, Wang XM, Li JX, Yang YJ, Lau JT, Hu YP (2006) Isolation and characterization of bipotent liver progenitor cells from adult mouse. Stem Cells 24:322–332
Marshall A, Rushbrook S, Davies SE, Morris LS, Scott IS, Vowler SL, Coleman N, Alexander G (2005) Relation between hepatocyte G1 arrest, impaired hepatic regeneration, and fibrosis in chronic hepatitis C virus infection. Gastroenterology 128:33–42
Mason D, Andre P, Bensussan A, Buckley C, Civin C, Clark E, Haas M de, Goyert S, Hadam M, Hart D, Horejsi V, Meuer S, Morrissey J, Schwartz-Albiez R, Shaw S, Simmons D, Uguccioni M, Schoot E van der, Vivier E, Zola H (2002) CD antigens 2001. Mod Pathol 15:71–76
Matas AJ, Sutherland DE, Steffes MW, Mauer SM, Sowe A, Simmons RL, Najarian JS (1976) Hepatocellular transplantation for metabolic deficiencies: decrease of plasms bilirubin in Gunn rats. Science 192:892–894
Michalopoulos GK, DeFrances MC (1997) Liver regeneration. Science 276:60–66
Michel JL, Rabier D, Rambaud C, Kamoun P, Brousse N, Vassault A, Pla M, Calise D, Revillon Y (1993) Intrasplenic transplantation of hepatocytes in spf-ash mice with congenital ornithine transcarbamylase deficiency. Chirurgie 119:666–671
Mitchell C, Fausto N (2002) Bone marrow-derived hepatocytes: rare but promising. Am J Pathol 161:349–350
Miyashita H, Suzuki A, Fukao K, Nakauchi H, Taniguchi H (2002) Evidence for hepatocyte differentiation from embryonic stem cells in vitro. Cell Transplant 11:429–434
Muraca M, Gerunda G, Neri D, Vilei MT, Granato A, Feltracco P, Meroni M, Giron G, Burlina AB (2002) Hepatocyte transplantation as a treatment for glycogen storage disease type 1a. Lancet 359:317–318
Newsome PN, Johannessen I, Boyle S, Dalakas E, McAulay KA, Samuel K, Rae F, Forrester L, Turner ML, Hayes PC, Harrison DJ, Bickmore WA, Plevris JN (2003) Human cord blood-derived cells can differentiate into hepatocytes in the mouse liver with no evidence of cellular fusion. Gastroenterology 124:1891–1900
Nierhoff D, Ogawa A, Oertel M, Chen YQ, Shafritz DA (2005) Purification and characterization of mouse fetal liver epithelial cells with high in vivo repopulation capacity. Hepatology 42:130–139
Overturf K, al-Dhalimy M, Ou CN, Finegold M, Grompe M (1997) Serial transplantation reveals the stem-cell-like regenerative potential of adult mouse hepatocytes. Am J Pathol 151:1273–1280
Parent R, Marion MJ, Furio L, Trepo C, Petit MA (2004) Origin and characterization of a human bipotent liver progenitor cell line. Gastroenterology 126:1147–1156
Petersen BE (2001) Hepatic “stem” cells: coming full circle. Blood Cells Mol Dis 27:590–600
Petersen BE, Goff JP, Greenberger JS, Michalopoulos GK (1998) Hepatic oval cells express the hematopoietic stem cell marker Thy-1 in the rat. Hepatology 27:433–445
Petersen BE, Bowen WC, Patrene KD, Mars WM, Sullivan AK, Murase N, Boggs SS, Greenberger JS, Goff JP (1999) Bone marrow as a potential source of hepatic oval cells. Science 284:1168–1170
Petersen BE, Grossbard B, Hatch H, Pi L, Deng J, Scott EW (2003) Mouse A6-positive hepatic oval cells also express several hematopoietic stem cell markers. Hepatology 37:632–640
Quintana-Bustamante O, Alvarez-Barrientos A, Kofman AV, Fabregat I, Bueren JA, Theise ND, Segovia JC (2006) Hematopoietic mobilization in mice increases the presence of bone marrow-derived hepatocytes via in vivo cell fusion. Hepatology 43:108–116
Rathjen J, Rathjen PD (2001) Mouse ES cells: experimental exploitation of pluripotent differentiation potential. Curr Opin Genet Dev 11:587–594
Reyes M, Lund T, Lenvik T, Aguiar D, Koodie L, Verfaillie CM (2001) Purification and ex vivo expansion of postnatal human marrow mesodermal progenitor cells. Blood 98:2615–2625
Roskams T, Yang SQ, Koteish A, Durnez A, DeVos R, Huang X, Achten R, Verslype C, Diehl AM (2003) Oxidative stress and oval cell accumulation in mice and humans with alcoholic and nonalcoholic fatty liver disease. Am J Pathol 163:1301–1311
Sakaida I, Terai S, Yamamoto N, Aoyama K, Ishikawa T, Nishina H, Okita K (2004) Transplantation of bone marrow cells reduces CCl4-induced liver fibrosis in mice. Hepatology 40:1304–1311
Sandhu JS, Petkov PM, Dabeva MD, Shafritz DA (2001) Stem cell properties and repopulation of the rat liver by fetal liver epithelial progenitor cells. Am J Pathol 159:1323–1334
Scharenberg CW, Harkey MA, Torok-Storb B (2002) The ABCG2 transporter is an efficient Hoechst 33342 efflux pump and is preferentially expressed by immature human hematopoietic progenitors. Blood 99:507–512
Schneider A, Attaran M, Meier PN, Strassburg C, Manns MP, Ott M, Barthold M, Arseniev L, Becker T, Panning B (2006) Hepatocyte transplantation in an acute liver failure due to mushroom poisoning. Transplantation 82:1115–1116
Schwartz RE, Reyes M, Koodie L, Jiang Y, Blackstad M, Lund T, Lenvik T, Johnson S, Hu WS, Verfaillie CM (2002) Multipotent adult progenitor cells from bone marrow differentiate into functional hepatocyte-like cells. J Clin Invest 109:1291–1302
Sharma AD, Cantz T, Richter R, Eckert K, Henschler R, Wilkens L, Jochheim-Richter A, Arseniev L, Ott M (2005) Human cord blood stem cells generate human cytokeratin 18-negative hepatocyte-like cells in injured mouse liver. Am J Pathol 167:555–564
Shi Z, Wakil AE, Rockey DC (1997) Strain-specific differences in mouse hepatic wound healing are mediated by divergent T helper cytokine responses. Proc Natl Acad Sci USA 94:10663–10668
Shimano K, Satake M, Okaya A, Kitanaka J, Kitanaka N, Takemura M, Sakagami M, Terada N, Tsujimura T (2003) Hepatic oval cells have the side population phenotype defined by expression of ATP-binding cassette transporter ABCG2/BCRP1. Am J Pathol 163:3–9
Sigal SH, Gupta S, Gebhard DF Jr, Holst P, Neufeld D, Reid LM (1995) Evidence for a terminal differentiation process in the rat liver. Differentiation 59:35–42
Sokal EM, Smets F, Bourgois A, Van Maldergem L, Buts JP, Reding R, Bernard Otte J, Evrard V, Latinne D, Vincent MF, Moser A, Soriano HE (2003) Hepatocyte transplantation in a 4-year-old girl with peroxisomal biogenesis disease: technique, safety, and metabolic follow-up. Transplantation 76:735–738
Soto-Gutierrez A, Kobayashi N, Rivas-Carrillo JD, Navarro-Alvarez N, Zhao D, Okitsu T, Noguchi H, Basma H, Tabata Y, Chen Y, Tanaka K, Narushima M, Miki A, Ueda T, Jun HS, Yoon JW, Lebkowski J, Tanaka N, Fox IJ (2006) Reversal of mouse hepatic failure using an implanted liver-assist device containing ES cell-derived hepatocytes. Nat Biotechnol 24:1412–1419
Strom SC, Fisher RA, Thompson MT, Sanyal AJ, Cole PE, Ham JM, Posner MP (1997) Hepatocyte transplantation as a bridge to orthotopic liver transplantation in terminal liver failure. Transplantation 63:559–569
Suzuki A, Zheng YW, Kaneko S, Onodera M, Fukao K, Nakauchi H, Taniguchi H (2002) Clonal identification and characterization of self-renewing pluripotent stem cells in the developing liver. J Cell Biol 156:173–184
Tan J, Hytiroglou P, Wieczorek R, Park YN, Thung SN, Arias B, Theise ND (2002) Immunohistochemical evidence for hepatic progenitor cells in liver diseases. Liver 22:365–373
Tanabe Y, Tajima F, Nakamura Y, Shibasaki E, Wakejima M, Shimomura T, Murai R, Murawaki Y, Hashiguchi K, Kanbe T, Saeki T, Ichiba M, Yoshida Y, Mitsunari M, Yoshida S, Miake J, Yamamoto Y, Nagata N, Harada T, Kurimasa A, Hisatome I, Terakawa N, Murawaki Y, Shiota G (2004) Analyses to clarify rich fractions in hepatic progenitor cells from human umbilical cord blood and cell fusion. Biochem Biophys Res Commun 324:711–718
Terai S, Ishikawa T, Omori K, Aoyama K, Marumoto Y, Urata Y, Yokoyama Y, Uchida K, Yamasaki T, Fujii Y, Okita K, Sakaida I (2006) Improved liver function in patients with liver cirrhosis after autologous bone marrow cell infusion therapy. Stem Cells 24:2292–2298
Teratani T, Yamamoto H, Aoyagi K, Sasaki H, Asari A, Quinn G, Sasaki H, Terada M, Ochiya T (2005) Direct hepatic fate specification from mouse embryonic stem cells. Hepatology 41:836–846
Theise ND, Badve S, Saxena R, Henegariu O, Sell S, Crawford JM, Krause DS (2000a) Derivation of hepatocytes from bone marrow cells in mice after radiation-induced myeloablation. Hepatology 31:235–240
Theise ND, Nimmakayalu M, Gardner R, Illei PB, Morgan G, Teperman L, Henegariu O, Krause DS (2000b) Liver from bone marrow in humans. Hepatology 32:11–16
Thorgeirsson SS (1996) Hepatic stem cells in liver regeneration. FASEB J 10:1249–1256
Thorgeirsson SS, Grisham JW (2006) Hematopoietic cells as hepatocyte stem cells: a critical review of the evidence. Hepatology 43:2–8
Tsanev R (1975) Cell cycle and liver function. In: Reinert J, Hottzer H (ed) Cell cycle and cell differentiation. Springer, Berlin Heidleberg New York, pp 197–248
Tsao MS, Smith JD, Nelson KG, Grisham JW (1984) A diploid epithelial cell line from normal adult rat liver with phenotypic properties of “oval” cells. Exp Cell Res 154:38–52
Tsuchiya A, Heike T, Baba S, Fujino H, Umeda K, Matsuda Y, Nomoto M, Ichida T, Aoyagi Y, Nakahata T (2007) Long-term culture of postnatal mouse hepatic stem/progenitor cells and their relative developmental hierarchy. Stem Cells 25:895–902
Turrini P, Monego G, Gonzalez J, Cicuzza S, Bonanno G, Zelano G, Rosenthal N, Paonessa G, Laufer R, Padron J (2005) Human hepatocytes in mice receiving pre-immune injection with human cord blood cells. Biochem Biophys Res Commun 326:66–73
Uchida N, Fujisaki T, Eaves AC, Eaves CJ (2001) Transplantable hematopoietic stem cells in human fetal liver have a CD34(+) side population (SP )phenotype. J Clin Invest 108:1071–1077
Vassilopoulos G, Wang PR, Russell DW (2003) Transplanted bone marrow regenerates liver by cell fusion. Nature 422:901–904
Vree JM de, Jacquemin E, Sturm E, Cresteil D, Bosma PJ, Aten J, Deleuze JF, Desrochers M, Burdelski M, Bernard O, Oude Elferink RP, Hadchouel M (1998) Mutations in the MDR3 gene cause progressive familial intrahepatic cholestasis. Proc Natl Acad Sci USA 95:282–287
Wagers AJ, Sherwood RI, Christensen JL, Weissman IL (2002) Little evidence for developmental plasticity of adult hematopoietic stem cells. Science 297:2256–2259
Wang X, Ge S, McNamara G, Hao QL, Crooks GM, Nolta JA (2003a) Albumin-expressing hepatocyte-like cells develop in the livers of immune-deficient mice that received transplants of highly purified human hematopoietic stem cells. Blood 101:4201–4208
Wang X, Willenbring H, Akkari Y, Torimaru Y, Foster M, Al-Dhalimy M, Lagasse E, Finegold M, Olson S, Grompe M (2003b) Cell fusion is the principal source of bone-marrow-derived hepatocytes. Nature 422:897–901
Wiederkehr JC, Kondos GT, Pollak R (1990) Hepatocyte transplantation for the low-density lipoprotein receptor-deficient state. A study in the Watanabe rabbit. Transplantation 50:466–471
Wiemann SU, Satyanarayana A, Tsahuridu M, Tillmann HL, Zender L, Klempnauer J, Flemming P, Franco S, Blasco MA, Manns MP, Rudolph KL (2002) Hepatocyte telomere shortening and senescence are general markers of human liver cirrhosis. FASEB J 16:935–942
Willenbring H, Bailey AS, Foster M, Akkari Y, Dorrell C, Olson S, Finegold M, Fleming WH, Grompe M (2004) Myelomonocytic cells are sufficient for therapeutic cell fusion in liver. Nat Med 10:744–748
Yamada T, Yoshikawa M, Kanda S, Kato Y, Nakajima Y, Ishizaka S, Tsunoda Y (2002) In vitro differentiation of embryonic stem cells into hepatocyte-like cells identified by cellular uptake of indocyanine green. Stem Cells 20:146–154
Yin Y, Lim YK, Salto-Tellez M, Ng SC, Lin CS, Lim SK (2002) AFP(+), ESC-derived cells engraft and differentiate into hepatocytes in vivo. Stem Cells 20:338–346
Yoshida Y, Tokusashi Y, Lee GH, Ogawa K (1996) Intrahepatic transplantation of normal hepatocytes prevents Wilson’s disease in Long-Evans cinnamon rats. Gastroenterology 111:1654–1660
Yovchev MI, Grozdanov PN, Joseph B, Gupta S, Dabeva MD (2007) Novel hepatic progenitor cell surface markers in the adult rat liver. Hepatology 45:139–149
Author information
Authors and Affiliations
Corresponding author
Appendix
Appendix

Stem cells in liver regeneration and therapy. Under physiological conditions, the division and proliferation of mature hepatocytes maintain hepatocyte number, i.e. liver mass. In humans and rodents, a 75% partial hepatectomy can be regenerated by this capacity. Under experimental conditions or carcinogenic changes, hepatocyte proliferation can be blocked. In this case, resident hepatic stem/progenitor cells (rHSPC) proliferate and differentiate into hepatocytes. The recent literature suggests the existence of an extra-hepatic stem cell pool that contributes to liver regeneration. Bone-marrow-cell-derived cells, e.g. mesenchymal stem cells, can give rise to hepatocytes in distinct experimental settings. Cell transplantation of adult and fetal hepatocytes has been established at the stage of phase-I clinical trials in patients with metabolic liver disorders or acute liver failure. Embryonic-stem-cell-derived hepatic cells are currently being investigated as a transplantable cell source in order to overcome limitations in cell number and tissue compatibility for future therapeutic strategies
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Cantz, T., Manns, M.P. & Ott, M. Stem cells in liver regeneration and therapy. Cell Tissue Res 331, 271–282 (2008). https://doi.org/10.1007/s00441-007-0483-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00441-007-0483-6