
Abstract
The proportion of never smoker non-small cell lung cancer (NSCLC) in Asia is about 30–40%. Despite the striking demographics and high prevalence of never smoker NSCLC, the exact causes still remain undetermined. Although several genome wide association (GWA) studies were conducted to find susceptibility loci for lung cancer in never smokers, no regions were replicated except for 5p15.33, suggesting locus heterogeneity and different environmental toxic effects. To identify genetic loci associated with susceptibility of lung cancer in never smokers, we performed a GWA analysis using the Affymetrix 6.0 SNP array. For discovery GWA set, we recruited 446 never smoking Korean patients with NSCLC and 497 normal subjects. We tested association of SNPs with lung cancer susceptibility using the Cochran-Armitage trend test. For validation, 39 SNPs were selected from the top 50 SNPs and five additional SNPs were selected in the DAB1 gene region which showed significant associations in the GWA analysis. The validation SNPs were genotyped in an independent sample including 434 patients and 1,000 controls. Among the 44 validation SNPs, two SNPs (rs11080466 and rs11663246) near the APCDD1, NAPG and FAM38B genes in the 18p11.22 region were replicated. P value of rs11080466 was 1.08 × 10−6 in the combined sets (2.68 × 10−5 in the discovery set and 2.60 × 10−3 in the validation set) and odds ratio was 0.68 (0.58–0.79). We observed similar association for rs11663246. Our result suggests the 18p11.22 region as a novel lung cancer susceptibility locus in never smokers.
Similar content being viewed by others

Introduction
Lung cancer is the leading cause of cancer death worldwide including Korea (Jee et al. 1998; Marugame and Hirabayashi 2009; Parkin et al. 2005). While cigarette smoking is the main cause of lung cancer, 10% of all patients with lung cancer worldwide are considered never smoker which is defined as less than 100 cigarettes in their lifetime (Subramanian and Govindan 2007). Especially, the proportion of never smokers in Asia including Korea is about 30–40% (Koo and Ho 1990), indicating other environmental factors might be involved in carcinogenesis of never smoker lung cancer.
Never smoker non-small cell lung cancer (NSCLC) has distinct clinical features and outcomes, in which the incidence of activating mutations in epidermal growth factor receptor (EGFR) tyrosine kinase is nearly 10 times higher than smokers, and dramatic response rate to EGFR tyrosine kinase inhibitors (Shigematsu et al. 2005). Recently, echinoderm microtubule-associated protein-like 4-anaplastic lymphoma kinase (EML4-ALK) fusion gene was commonly found in never smokers (Soda et al. 2007). So far, these two molecular pathways account for 40–50% of cases of lung cancer in never smokers.
Despite the striking demographics and high prevalence of never smoker NSCLC, the exact causes still remain undetermined. Until now, several factors, such as second-hand tobacco smoke, radon exposure, environmental pollutants, and cooking oil fumes have been considered as the major causes (Brennan et al. 2004; Darby et al. 2005; Lan et al. 2002). Given that, it can be hypothesized that there might be a genetic susceptibility to develop lung cancer even after trivial exposure to environmental carcinogen or toxin.
Recently, genome-wide association (GWA) studies have demonstrated that three human genomic regions at chromosomes 5p15, 15q25, and 6p21 are associated with susceptibility to lung cancer in European and American populations (Amos et al. 2008; Hung et al. 2008; McKay et al. 2008; Rafnar et al. 2009; Spitz et al. 2008). The 15q25 region encoding nicotinic acetylcholine receptor subunit genes (CHRNA5, CHRNA3, and CHRNB4) was associated with the risk of lung cancer and also considered to be related to nicotine dependence in smokers. Another 6p21 chromosome region was associated with risk of lung cancer. The third region at 5p15 contains two genes; the human telomerase reverse transcriptase gene (TERT) and cleft lip and palate transmembrane 1-like gene (CLPTM1L). The replication study with a small series of never smoker lung cancer has shown statistically significant association between lung cancer risk and 5p15.33 genotypes (Wang et al. 2008). Another GWA study of lung adenocarcinoma in never smoking Asian women also showed that common genetic variants in the TERT-CLPTM1L locus on chromosome 5p15.33 are associated with risk for lung adenocarcinoma (Hsiung et al. 2010). However, the other GWA study of never smoker lung cancer in mostly American population demonstrated that novel genetic variants at 13q31.3 are associated with susceptibility to never smoker lung cancer but the genetic variants at 5p15.33 was not replicated in this study (Li et al. 2010).
Although several studies were conducted to find susceptibility loci for lung cancer in never smokers, no regions were replicated except for 5p15.33, suggesting locus heterogeneity and different environmental toxic effects. In this study, to identify genetic loci associated with susceptibility of lung cancer in never smokers in Korea, we conducted a genome-wide association analysis using the Affymetrix 6.0 single nucleotide polymorphism (SNP) array (Affymetrix, Inc., Santa Clara, CA).
Materials and methods
Study population
For discovery set, a total of 446 patients who were never smokers and diagnosed with histologically confirmed NSCLC at Samsung Medical Center were enrolled in this study. Never smokers were defined as individuals who had smoked less than 100 cigarettes during their lifetime. Four hundred and ninety seven control subjects were recruited from the Korea Association REsource (KARE) project, which has prospectively collected more than 10,000 subjects and has been described in a previous study (Cho et al. 2009). The control subjects can be regarded as healthy individuals because patients with an apparent disease status were excluded from the cohort.
For independent validation set, a total of 434 patients and 1,000 control subjects were collected. Two hundred cases were obtained from Kyungpook National University Hospital (KNUH), 175 from Korea University Medical Center (KUMC), and 59 from Samsung Medical Center (SMC). Written informed consent was obtained from all patients at each of the participating institutions. Research protocol was approved by the institutional review boards at each institution. Individuals who serve as control were obtained from the KARE project.
Genotyping and quality control
An ethylenediamine tetraacetic acid (EDTA) venous blood was collected from the registered patients. Single nucleotide polymorphism (SNP) genotyping was performed using the Affymetrix Genome-Wide Human SNP Array 6.0 which includes more than 906,600 SNPs (Affymetrix, Inc., Santa Clara, CA). Before genotyping, we determined yields of pure double stranded genomic DNA using the Wizard Genomic DNA Purification Kit (Promega, Corp., Madison, WI). Samples were normalized to 50 ng/μl and the normalized genomic DNA (5 μl) from each sample was used as a template for the Affymetrix 6.0 Assays. Genotyping reactions were performed using the Affymetrix Genome-Wide Human SNP Nsp/Sty 5.0/6.0 kit reagents and protocols. Genotypes were called by the Birdseed algorithm of the Affymetrix Genotyping Console version 3.0.2. All procedures were done according to recommended protocols of the manufacturer. After excluding 174,617 SNPs of call rate <99%, 273,926 SNPs of minor allele frequency (MAF) <5%, and 6,532 SNPs showing deviation from Hardy–Weinberg equilibrium (P < 0.001) in controls, 474,503 autosomal SNPs were analyzed. Two patient samples with call rate <95% were excluded. Candidate validation SNPs selected from the discovery analysis were genotyped in the validation sample using the MassARRAY® system (Sequenom, Inc., San Diego, CA).
Statistical analysis
The population structures of our samples were examined to confirm genetic homogeneity and assess stratification using the multidimensional scaling (MDS) method. Affymetrix 6.0 data for East Asian (JPT + CHB), Caucasian (CEU) and African (YRI) populations from the International HapMap Project were used for MDS analysis. Genomic inflation factor (λ) was calculated based on median Chi-squared statistic. An association between each SNP and lung cancer susceptibility was tested using allelic test with the Cochran-Armitage trend test. Among the top 50 SNPs, SNPs that showed erroneous genotype clustering patterns were excluded by visual inspection. For validation, 39 SNPs that passed visual inspection from the top 50 SNPs were selected and five additional SNPs (P < 0.001) were selected in the DAB1 gene region to increase coverage of this region because the peak of the region seemed promising at discovery stage. A total of 44 SNPs were genotyped. All statistical analyses were conducted using the PLINK 1.06 (Purcell et al. 2007), and R 2.9.1 software. Linkage disequilibrium (LD) structure was assessed and SNP tagging was conducted using HaploView version 4.1 (Barrett et al. 2005). We also performed SNP imputation to increase genome-wide coverage for further analyses. IMPUTE program version 1.0.0 was used to impute additional polymorphic SNPs that were not covered by the Affymetrix 6.0 array (Howie et al. 2009). The reference panel used for imputation was composed of 90 known JPT + CHB haplotypes from the International HapMap Project data (Phase II Public Release #22 NCBI Build 36).
Results
Study population description and clinical characteristics
In order to identify common genetic variants associated with never smoker NSCLC, we finally conducted a GWA analysis using a total of 878 lung cancer patients and 1,497 healthy normal control subjects. Due to the high proportion of never smokers in women than in men, the proportion of male patients was low compared to control subjects (48.9% in discovery controls and 58.5% in validation controls) from the KARE cohort (Table 1). We observed no genetic substructure within study populations while there was a partial distinction between the study populations and the International HapMap East Asian (JPT + CHB) populations (Supplementary Fig. 1). The most frequent histologic type in all the cases was adenocarcinoma.
Association in the discovery set
In a discovery stage, we evaluated 474,503 common SNPs (MAF > 5%). The MDS analysis demonstrated that the genetic variations exhibited by our Korean subjects overlap with those from JPT + CHB and are clearly distinct from CEU and YRI according to the International HapMap Project data (Supplementary Fig. 2). The distributions of observed P values for association tests across all SNPs tested showed no evidence of overall systematic bias (λ = 1.042) from the expected P values, and the excess of low P values was consistent with the presence of true associations (quantile–quantile plot; Supplementary Fig. 3). These observations indicate that our samples are genetically homogeneous and the associations will be attributable to genetic difference of lung cancer susceptibility.
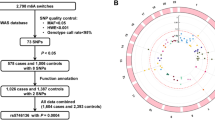
In the GWA analysis for 444 cases and 497 controls, we selected top 50 SNPs (Cochran-Armitage trend test; Supplementary Table 1). Manhattan plot of genome-wide association results illustrated clusters of significant association peaks in several regions (Fig. 1). Approximately 20 annotated genes, including possible cancer-related candidate genes, CCND2, DAB1, FRK, HDAC9, and IKBKAP, were located on or nearby these loci. A remarkable association peak was observed nearby the DAB1 gene on 1p32.2, where several consecutive SNPs show low P values.

Manhattan plot of genome-wide association results. P values were calculated using the Cochran-Armitage trend test. The validated region, 18p11.22, in the replication set was presented (see Fig. 2 for details)
The association of 18p11.22 was replicated in a different validation set
Among the top 50 SNPs, we excluded 11 SNPs with bad clustering through visual inspection and performed a validation study using the remaining 39 SNPs in another set consisting of Korean lung cancer patients (434 cases and 1,000 controls) recruited from KNUH, KUMC and SMC. While the association of 1p32.2 and most other loci in the discovery set was not replicated in the validation set, two SNPs, rs11080466 and rs11663246, in 18p11.22 showed a significant association in the validation set (P = 2.60 × 10−3 and 6.34 × 10−3, respectively) (Table 2). They were even more significant when analyzed in combination with the discovery set (P = 1.08 × 10−6 and 2.32 × 10−6, respectively). When patients with lung adenocarcinoma were analyzed, the association was still significant (Table 2). The two replicated SNPs were in the same LD block (r 2 = 0.987 and D′ = 1). The regional association plot of the 18p11.22 region in the original discovery set illustrated that the significant peak was located in intron of the FAM38B gene (Fig. 2). The 27 SNPs with P values < 0.05 and their tagging SNPs in the three HapMap populations are listed in Supplementary Table 2. For rs11080466, the allele C was underrepresented in cases when compared to controls with an odds ratio (OR) of 0.68 (95% confidence interval [CI], 0.58–0.79; Table 2) in the combined set. For rs11663246, the minor T allele was underrepresented in cases with an OR of 0.69 (95% CI, 0.59–0.80). The MAF of rs11080466 was consistently low in cases regardless of their origin (Fig. 3).

Regional association plot of the 18p11.22 region. Manhattan plot of genome-wide association results. P values were calculated using the Cochran-Armitage trend test. Filled circles and gray shaded circles indicate genotyped SNPs and imputed SNPs in the discovery set, respectively. Recombination rates are shown as a gray line and were estimated using the HapMap combined data. Arrows indicate the locations of genes. The lower panel presents an LD map based on r 2 values computed using the HapMap JPT + CHB data

Minor allele frequency of rs11080466 for each study data. SMC Samsung Medical Center, KNUH Kyungpook National University Hospital, KUMC Korea University Medical Center
Discussion
We found a novel genetic variant locus at 18p11.22 which has not been identified in previous GWA studies of never smoker NSCLC. Recent GWA study of lung adenocarcinoma in never smoking Asian women including 594 cases demonstrated that common genetic variants in the TERT-CLPTM1L locus on chromosome 5p15.33 are associated with risk for lung adenocarcinoma (Hsiung et al. 2010). This finding was independently replicated from East Asia totaling 1,164 lung adenocarcinoma and 1,736 controls, which contains substantially large sample size. The combined replication study confirmed that rs2736100 was associated with risk for lung cancer with P = 5.38 × 10−11 and allelic OR = 1.44. Given that, we tried to confirm the association with our cohort. However, in this study, we only observed P value of 0.008 (top 1% rank, 4,842nd among 474,503 SNPs) for rs2736100 at 5p15.33. It is of note that the results of Korean population did not show significant association for the rs2736100 SNP in the previous study (Hsiung et al. 2010), which is consistent with our result. Although rs2736100 did not reach a genome-wide significance level in our data, allelic frequency pattern of the Korean data (G allele frequency of 0.44 in cases and 0.38 in controls; allelic OR of 1.29 [95% CI, 1.07–1.55]) showed a similar trend with that of Hsiung et al.’s (2010) data (G allele frequency of 0.48 in cases and 0.39 in controls). Therefore, we could not exclude 5p15.33 as a potential risk locus because inconsistent results between the Korean and Chinese populations might be attributed to genetic heterogeneity between the populations, small number of samples in the Korean cohorts and/or different environmental predisposing factors. Moreover, another GWA study of never smoker lung cancer in mostly American population also demonstrated novel genetic variants at 13q31.3 as susceptibility to lung cancer in never smokers which has not been reported yet. However, the genetic variants at 5p15.33 were not replicated in this study (Li et al. 2010). All together, these inconsistent observations might be attributed to the ethnic difference even in East Asian population. It could be suggested that there is a large heterogeneity in genetic background as well as in environmental predisposing factors among different ethnic groups with regard to the susceptibility to NSCLC in never smokers.
Another possible explanation can be the different environmental toxic agents and exposure to second hand smoke. It has long been considered that cooking oil fumes are associated with increased lung cancer risk in Asian never smokers, especially China and Taiwan (Hosgood et al. 2007). However, the relevance of the difference of indoor air pollution via cooking methods amongst different Asian countries with 18p11.22 is not clear, and thus the exact functional relevance of 18p11.22 with tumorigenesis and environmental toxic agents still needs to be further examined.
We identified 50 candidate SNPs. Although this study has limitation due to the lack of information about potential confounding variables such as age, sex, exposure to second-hand smoke, or family history of cancer, two of these candidate SNPs, rs11080466 and rs11663246 were replicated in validation set. These two SNPs are in linkage disequilibrium and located in intron of the FAM38B gene at 18p11.22. Our data showed a relatively weak association of the locus, and this might be attributed to the small number of subjects and modest impact of the responsible gene. Recently, it has been reported that FAM38B (Piezo2) along with FAM38A (Piezo1) are vertebrate multipass transmembrane proteins with homologs in invertebrates, plants, and protozoa and considered as essential components of distinct mechanically activated cation channels (Coste et al. 2010). FAM38B is expressed in various tissues and has potential role in touch and pain sensation, leading to a broad role in mechanotransduction (Coste et al. 2010). The association of FAM38B and cancer has not been explored, but unpublished data suggest that this gene might play a role in the tumorigenesis of lung cancer (Sethi 2008). Nonetheless, the exact role of FAM38B in tumorigenesis still remains largely unknown, and further biological studies would be needed.
In summary, we have identified a novel genetic locus at 18p11.22 region which is associated with susceptibility of never smoker NSCLC in Korean populations. We failed to replicate findings of previous GWA studies which may indicate a large genetic heterogeneity in the genetics of never smoker NSCLC. Further replication studies in larger populations are necessary to clarify our hypothesis.
References
Amos CI, Wu X, Broderick P, Gorlov IP, Gu J, Eisen T, Dong Q, Zhang Q, Gu X, Vijayakrishnan J, Sullivan K, Matakidou A, Wang Y, Mills G, Doheny K, Tsai YY, Chen WV, Shete S, Spitz MR, Houlston RS (2008) Genome-wide association scan of tag SNPs identifies a susceptibility locus for lung cancer at 15q25.1. Nat Genet 40:616–622
Barrett JC, Fry B, Maller J, Daly MJ (2005) Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 21:263–265
Brennan P, Buffler PA, Reynolds P, Wu AH, Wichmann HE, Agudo A, Pershagen G, Jockel KH, Benhamou S, Greenberg RS, Merletti F, Winck C, Fontham ET, Kreuzer M, Darby SC, Forastiere F, Simonato L, Boffetta P (2004) Secondhand smoke exposure in adulthood and risk of lung cancer among never smokers: a pooled analysis of two large studies. Int J Cancer 109:125–131
Cho YS, Go MJ, Kim YJ, Heo JY, Oh JH, Ban HJ, Yoon D, Lee MH, Kim DJ, Park M, Cha SH, Kim JW, Han BG, Min H, Ahn Y, Park MS, Han HR, Jang HY, Cho EY, Lee JE, Cho NH, Shin C, Park T, Park JW, Lee JK, Cardon L, Clarke G, McCarthy MI, Lee JY, Oh B, Kim HL (2009) A large-scale genome-wide association study of Asian populations uncovers genetic factors influencing eight quantitative traits. Nat Genet 41:527–534
Coste B, Mathur J, Schmidt M, Earley TJ, Ranade S, Petrus MJ, Dubin AE, Patapoutian A (2010) Piezo1 and Piezo2 are essential components of distinct mechanically activated cation channels. Science 330:55–60
Darby S, Hill D, Auvinen A, Barros-Dios JM, Baysson H, Bochicchio F, Deo H, Falk R, Forastiere F, Hakama M, Heid I, Kreienbrock L, Kreuzer M, Lagarde F, Makelainen I, Muirhead C, Oberaigner W, Pershagen G, Ruano-Ravina A, Ruosteenoja E, Rosario AS, Tirmarche M, Tomasek L, Whitley E, Wichmann HE, Doll R (2005) Radon in homes and risk of lung cancer: collaborative analysis of individual data from 13 European case-control studies. BMJ 330:223
Hosgood HD 3rd, Berndt SI, Lan Q (2007) GST genotypes and lung cancer susceptibility in Asian populations with indoor air pollution exposures: a meta-analysis. Mutat Res 636:134–143
Howie BN, Donnelly P, Marchini J (2009) A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet 5:e1000529
Hsiung CA, Lan Q, Hong YC, Chen CJ, Hosgood HD, Chang IS, Chatterjee N, Brennan P, Wu C, Zheng W, Chang GC, Wu T, Park JY, Hsiao CF, Kim YH, Shen H, Seow A, Yeager M, Tsai YH, Kim YT, Chow WH, Guo H, Wang WC, Sung SW, Hu Z, Chen KY, Kim JH, Chen Y, Huang L, Lee KM, Lo YL, Gao YT, Liu L, Huang MS, Jung TH, Jin G, Caporaso N, Yu D, Kim CH, Su WC, Shu XO, Xu P, Kim IS, Chen YM, Ma H, Shen M, Cha SI, Tan W, Chang CH, Sung JS, Zhang M, Yang TY, Park KH, Yuenger J, Wang CL, Ryu JS, Xiang Y, Deng Q, Hutchinson A, Kim JS, Cai Q, Landi MT, Yu CJ, Tucker M, Hung JY, Lin CC, Perng RP, Boffetta P, Chen CY, Chen KC, Yang SY, Hu CY, Chang CK, Fraumeni JF, Jr., Chanock S, Yang PC, Rothman N, Lin D (2010) The 5p15.33 locus is associated with risk of lung adenocarcinoma in never-smoking females in Asia. PLoS Genet 6:e1001051
Hung RJ, McKay JD, Gaborieau V, Boffetta P, Hashibe M, Zaridze D, Mukeria A, Szeszenia-Dabrowska N, Lissowska J, Rudnai P, Fabianova E, Mates D, Bencko V, Foretova L, Janout V, Chen C, Goodman G, Field JK, Liloglou T, Xinarianos G, Cassidy A, McLaughlin J, Liu G, Narod S, Krokan HE, Skorpen F, Elvestad MB, Hveem K, Vatten L, Linseisen J, Clavel-Chapelon F, Vineis P, Bueno-de-Mesquita HB, Lund E, Martinez C, Bingham S, Rasmuson T, Hainaut P, Riboli E, Ahrens W, Benhamou S, Lagiou P, Trichopoulos D, Holcatova I, Merletti F, Kjaerheim K, Agudo A, Macfarlane G, Talamini R, Simonato L, Lowry R, Conway DI, Znaor A, Healy C, Zelenika D, Boland A, Delepine M, Foglio M, Lechner D, Matsuda F, Blanche H, Gut I, Heath S, Lathrop M, Brennan P (2008) A susceptibility locus for lung cancer maps to nicotinic acetylcholine receptor subunit genes on 15q25. Nature 452:633–637
Jee SH, Kim IS, Suh I, Shin D, Appel LJ (1998) Projected mortality from lung cancer in South Korea, 1980–2004. Int J Epidemiol 27:365–369
Koo LC, Ho JH (1990) Worldwide epidemiological patterns of lung cancer in nonsmokers. Int J Epidemiol 19(Suppl 1):S14–S23
Lan Q, Chapman RS, Schreinemachers DM, Tian L, He X (2002) Household stove improvement and risk of lung cancer in Xuanwei, China. J Natl Cancer Inst 94:826–835
Li Y, Sheu CC, Ye Y, de Andrade M, Wang L, Chang SC, Aubry MC, Aakre JA, Allen MS, Chen F, Cunningham JM, Deschamps C, Jiang R, Lin J, Marks RS, Pankratz VS, Su L, Sun Z, Tang H, Vasmatzis G, Harris CC, Spitz MR, Jen J, Wang R, Zhang ZF, Christiani DC, Wu X, Yang P (2010) Genetic variants and risk of lung cancer in never smokers: a genome-wide association study. Lancet Oncol 11:321–330
Marugame T, Hirabayashi Y (2009) Comparison of time trends in lung cancer mortality (1990–2006) in the world, from the WHO Mortality Database. Jpn J Clin Oncol 39:696–697
McKay JD, Hung RJ, Gaborieau V, Boffetta P, Chabrier A, Byrnes G, Zaridze D, Mukeria A, Szeszenia-Dabrowska N, Lissowska J, Rudnai P, Fabianova E, Mates D, Bencko V, Foretova L, Janout V, McLaughlin J, Shepherd F, Montpetit A, Narod S, Krokan HE, Skorpen F, Elvestad MB, Vatten L, Njolstad I, Axelsson T, Chen C, Goodman G, Barnett M, Loomis MM, Lubinski J, Matyjasik J, Lener M, Oszutowska D, Field J, Liloglou T, Xinarianos G, Cassidy A, Vineis P, Clavel-Chapelon F, Palli D, Tumino R, Krogh V, Panico S, Gonzalez CA, Ramon Quiros J, Martinez C, Navarro C, Ardanaz E, Larranaga N, Kham KT, Key T, Bueno-de-Mesquita HB, Peeters PH, Trichopoulou A, Linseisen J, Boeing H, Hallmans G, Overvad K, Tjonneland A, Kumle M, Riboli E, Zelenika D, Boland A, Delepine M, Foglio M, Lechner D, Matsuda F, Blanche H, Gut I, Heath S, Lathrop M, Brennan P (2008) Lung cancer susceptibility locus at 5p15.33. Nat Genet 40:1404–1406
Parkin DM, Bray F, Ferlay J, Pisani P (2005) Global cancer statistics, 2002. CA Cancer J Clin 55:74–108
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC (2007) PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81:559–575
Rafnar T, Sulem P, Stacey SN, Geller F, Gudmundsson J, Sigurdsson A, Jakobsdottir M, Helgadottir H, Thorlacius S, Aben KK, Blondal T, Thorgeirsson TE, Thorleifsson G, Kristjansson K, Thorisdottir K, Ragnarsson R, Sigurgeirsson B, Skuladottir H, Gudbjartsson T, Isaksson HJ, Einarsson GV, Benediktsdottir KR, Agnarsson BA, Olafsson K, Salvarsdottir A, Bjarnason H, Asgeirsdottir M, Kristinsson KT, Matthiasdottir S, Sveinsdottir SG, Polidoro S, Hoiom V, Botella-Estrada R, Hemminki K, Rudnai P, Bishop DT, Campagna M, Kellen E, Zeegers MP, de Verdier P, Ferrer A, Isla D, Vidal MJ, Andres R, Saez B, Juberias P, Banzo J, Navarrete S, Tres A, Kan D, Lindblom A, Gurzau E, Koppova K, de Vegt F, Schalken JA, van der Heijden HF, Smit HJ, Termeer RA, Oosterwijk E, van Hooij O, Nagore E, Porru S, Steineck G, Hansson J, Buntinx F, Catalona WJ, Matullo G, Vineis P, Kiltie AE, Mayordomo JI, Kumar R, Kiemeney LA, Frigge ML, Jonsson T, Saemundsson H, Barkardottir RB, Jonsson E, Jonsson S, Olafsson JH, Gulcher JR, Masson G, Gudbjartsson DF, Kong A, Thorsteinsdottir U, Stefansson K (2009) Sequence variants at the TERT-CLPTM1L locus associate with many cancer types. Nat Genet 41:221–227
Sethi T (2008) http://talks.cam.ac.uk/talk/index/13412
Shigematsu H, Lin L, Takahashi T, Nomura M, Suzuki M, Wistuba II, Fong KM, Lee H, Toyooka S, Shimizu N, Fujisawa T, Feng Z, Roth JA, Herz J, Minna JD, Gazdar AF (2005) Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. J Natl Cancer Inst 97:339–346
Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S, Fujiwara S, Watanabe H, Kurashina K, Hatanaka H, Bando M, Ohno S, Ishikawa Y, Aburatani H, Niki T, Sohara Y, Sugiyama Y, Mano H (2007) Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 448:561–566
Spitz MR, Amos CI, Dong Q, Lin J, Wu X (2008) The CHRNA5–A3 region on chromosome 15q24–25.1 is a risk factor both for nicotine dependence and for lung cancer. J Natl Cancer Inst 100:1552–1556
Subramanian J, Govindan R (2007) Lung cancer in never smokers: a review. J Clin Oncol 25:561–570
Wang Y, Broderick P, Webb E, Wu X, Vijayakrishnan J, Matakidou A, Qureshi M, Dong Q, Gu X, Chen WV, Spitz MR, Eisen T, Amos CI, Houlston RS (2008) Common 5p15.33 and 6p21.33 variants influence lung cancer risk. Nat Genet 40:1407–1409
Acknowledgments
This work was supported by grants from Ministry of Health and Welfare, Republic of Korea (A040041) and Samsung Biomedical Research Institute, Republic of Korea (C-H7-103), by grants of the Korea Healthcare Technology R&D Project, Ministry of Health and Welfare, Republic of Korea (A070001 and A092255), by a grant of the Korea Health 21 R&D Project, Ministry of Health and Welfare, Republic of Korea (0405-MN01-0604-0007), by a grant of IN-SUMG Foundation for Medical Research (CA98721), and by a grant from Samsung Biomedical Research Institute, Seoul, Republic of Korea (C-H1-103).
Conflict of interest
The authors have nothing to disclose.
Author information
Authors and Affiliations
Corresponding authors
Additional information
M.-J. Ahn, H.-H. Won and J. Lee contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Ahn, MJ., Won, HH., Lee, J. et al. The 18p11.22 locus is associated with never smoker non-small cell lung cancer susceptibility in Korean populations. Hum Genet 131, 365–372 (2012). https://doi.org/10.1007/s00439-011-1080-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-011-1080-z