
Abstract
The rapid advancement of genome editing technologies has opened up new possibilities in the field of medicine. Nuclease-based techniques such as the CRISPR/Cas9 system are now used to target genetically linked disorders that were previously hard-to-treat. The CRISPR/Cas9 gene editing approach wields several advantages over its contemporary editing systems, notably in the ease of component design, implementation and the option of multiplex genome editing. While results from the early phase clinical trials have been encouraging, the small patient population recruited into these trials hinders a conclusive assessment on the safety aspects of the CRISPR/Cas9 therapy. Potential safety concerns include the lack of fidelity in the CRISPR/Cas9 system which may lead to unintended DNA modifications at non-targeted gene loci. This review focuses modifications to the CRISPR/Cas9 components that can mitigate off-target effects in in vitro and preclinical models and its translatability to gene therapy in patient populations.
Similar content being viewed by others


Introduction
The possibility of editing the genome of an organism, inserting, deleting or modifying DNA sequences at targeted loci, holds great promise for the advancement of gene therapy in clinical populations. In 1990, the FDA approved the very first US-based gene therapy-related clinical trial in which 2 children afflicted with ADA-linked severe combined immuno-deficiency (ADA−SCID) were infused with T-cells that had a corrected copy of the ADA gene [1]. Both patients responded positively to the treatment, with tests showing an increased number of functional T-cells that were able to survive for over a year following the last infusion. However, the lymphocytic ADA enzyme levels in one patient did not differ significantly after treatment owing to the low efficiency of the transgene integration via homologous recombination (HR). In addition, it was difficult to determine the exact contributions of the gene therapy to the beneficial results given that the patients were continued on PEG-ADA treatments concurrently [1]. Consequentially, clinical trials over the subsequent 30 years have shifted towards using next-generation engineered endonuclease-based gene editing technologies with a focus on higher efficiency and specificity. These include, but are not limited to, zinc-finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs) and clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 endonucleases. Of the three, ZFN and TALEN are nucleases containing a customisable DNA-binding domain, which is commonly fused to a FokI DNA-cleavage domain [2,3,4]. The CRISPR/Cas9 system consists of a DNA-cleaving endonuclease (Cas9) associated with a guide RNA that recognises and binds to the targeted sequences [5,6,7]. Regardless of the technique, the concept underlying gene editing with endonucleases is to induce a double-strand break (DSB) at targeted sites in the genomic DNA of the host cell, following which, DNA repair then proceeds via (1) the more predominant non-homologous end joining (NHEJ) that introduces a loss-of-function mutation through a reading frame shift or a premature stop codon or (2) a directed HR process that integrates transgene sequences into the genomic DNA through a supplied repair template [8]. These newer techniques demonstrated great therapeutic potential in ex vivo and in vivo preclinical models, which led to the approval of clinical trials in the USA that utilised these gene editing tools to ameliorate disease phenotypes [9,10,11,12].
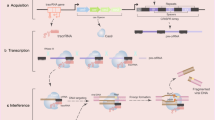
The results from the early phase clinical trials have so far been promising. However, given the small patient populations recruited into these trials, the safety of these gene therapies has yet to be fully evaluated. Specifically, potential off-target effects (OTEs) of these techniques have been highlighted in several studies demonstrating off-target ZFN and TALEN activity in in vitro and in vivo preclinical models [13,14,15,16,17]. Similarly, significant rates of OTEs had been reported in in vitro CRISPR/Cas9 edited human cell lines [18,19,20,21,22] and non-viable human embryos [23]. The CRISPR/Cas9 gene editing technique holds many advantages over ZFNs and TALENs in targeting disorders with a distinct genetic aetiology—the ease of designing the guide RNA sequences, the computational determination of OTEs based on genomic sequences with high similarity to the target locus (first-order sequence screens) [24,25,26,27] and the possibility of editing multiple genomic loci simultaneously [4]. Nonetheless, it remains prudent for clinical applications of CRISPR/Cas9 to implement constructs that can minimise OTEs. This current review focuses on modifications to the design of the CRISPR/Cas9 gene editing tool to mitigate OTEs and the subsequent delivery of CRISPR/Cas9 to the desired gene loci/cell populations in vivo to achieve effective gene editing clinically (Fig. 1).

Graphical outline of the review article and overview of the mitigating techniques that can be applied to minimise OTEs in CRISPR/Cas9 genome editing
The CRISPR/Cas9 system
The CRISPR/Cas9 gene editing tool was developed from the endogenous prokaryotic immune system in which DNA sequences (CRISPR sequences or spacers) from invading viruses and phages incorporate into the bacteria or archaea as clustered interrupted repeats [28,29,30]. Together with the Cas endonuclease, these CRISPR sequences are used to detect and cleave DNA from subsequent infections from the same virus [31, 32]. The CRISPR/Cas system is divided into 2 classes, with class 1 consisting of types I, III and IV Cas proteins while class 2 consists types II, V and VI Cas proteins [33]. The system is further subdivided into 19 subtypes based on the type of Cas protein [33]. In the class I system, multiple Cas proteins form an effective complex to cleave foreign DNA sequences while only a single Cas protein is required to do so in the class II system [34]. The CRISPR/Cas9 system falls under the class 2 type II classification [33] and the system derived from the Streptococcus pyogenes bacteria is the most extensively studied of the Cas9 proteins [35].
Activation of the CRISPR system is initiated by the transcription of the repeat-spacer sequences into precursor CRISPR RNA (pre-crRNA) [36]. Together with RNase III and the Cas9 endonuclease, the pre-crRNA is processed into mature crRNA by a trans-activating crRNA (tracrRNA) hybridised at the 5′ end to the pre-crRNA [5, 37]. The mature crRNA-tracrRNA product then associates with the Cas9 endonuclease to form an RNA-endonuclease complex [5]. In addition to directing the processing of pre-crRNA, the tracrRNA is also critical in maintaining the Cas9 protein in the activated state [38]. The Cas9 protein searches the genomic DNA for the presence of protospacer adjacent motifs (PAMs) [39]—short DNA sequences recognised by the CRISPR/Cas system as foreign genomic material [40]. In S. pyogenes, the canonical 5′-NGG PAM sequence (Fig. 2a) lies downstream of the target site [5]. Once detected, S. pyogenes Cas9 (SpCas9) binds to the appropriate PAM sequence and relaxes the crRNA-tracrRNA structure to allow crRNA to scan for complementary DNA sequences [39]. In this way, the crRNA functions as a guide to bring Cas9 to the target site. Hybridisation of crRNA to the matching target sequence then induces a conformational change which activates the nuclease domains in Cas9 to cleave the target site [41].

Stagewise schematic representations of target site recognition in CRISPR/Cas9-mediated genome editing with modifications to the sgRNA and Cas9 endonuclease to reduce OTEs. a The Cas9 endonuclease first scans the genomic DNA and binds to canonical PAM sequences (I). This induces a structural change in the sgRNA that allows the guide sequence to search and hybridise to complementary target sites upstream of the PAM (II). sgRNA-DNA hybridisation activates the Cas9 nuclease domains which then cleaves both strands of DNA (III). b sgRNA can be truncated at the 5′-end by 2–3 nucleotides or c modified at the 5′-end to contain 2 guanine nucleotides to improve the specificity of the guide sequence. d Cas9 nickase with only 1 active catalytic domain can be paired and e catalytically deactivated Cas9 fused to FokI nuclease that requires dimerisation for nuclease activity can be used to minimise off-target indels (insertion/deletion). f Base editors that convert a single cytosine base to thymine without requiring DSBs are less promiscuous at off-target sites. g Cas9 orthologs from other bacteria such as SaCas9 and h other Cas nucleases such as Cas12a that recognises alternative PAMs can be used to target novel DNA sequences and improve specificity
The CRISPR/Cas9 system was modified for more efficient endeavours through the fusion of the 3′-end of crRNA to the 5′-end of tracrRNA to generate a single-guide RNA (sgRNA) that contained the target recognition domain of crRNA and a hairpin loop that mimicked endogenous interactions between tracrRNA and crRNA [5].
Enhancing the fidelity of the CRISPR/Cas9 editing tool
Specificity of the CRISPR/Cas9 technique is a major concern in both preclinical and patient studies. Minimising OTEs while retaining/enhancing the rates of on-target activity of CRISPR/Cas9 is a challenge that involves DNA editing at the desired loci of the host genome, successful delivery of the CRISPR/Cas9 payload into the nucleus of the targeted cell populations, and in the case of in vivo gene editing, the correct tissues/organs. The techniques discussed in this section describe modifications to the sgRNA, Cas9 endonuclease and delivery modality of the CRISPR/Cas9 system with the common goal of eliminating DSBs at non-targeted gene loci (Fig. 2).
Designing and modifying the sgRNA
Selecting an appropriate sgRNA for the targeted DNA sequence is a crucial first step in avoiding OTEs. Many sgRNA design tools are available online that provide on-target and off-target predictions based on custom algorithms that may be species- and/or nuclease-specific. Factors such as the location of the cleave site within the gene, guanine content, counts and positions of mismatches between the sgRNA and protospacer sequence and non-canonical PAM sequences are duly taken into account in the models’ algorithms. A list of currently available sgRNA design web tools have been summarised by Cui et al. in a recent review article [42].
Conventional sgRNAs are designed to contain 20 nucleotides which are complementary to the target DNA sequence since longer sgRNA are found to be less effective [5, 6, 43,44,45]. However, Cas9 is able to bind to the target with a tolerance of up to 10 mismatches between the target sequence and the sgRNA while DNA cleavage can be detected at a small minority of loci with a mismatch of 3–5 base pairs [18, 20, 46]. To overcome this issue, the 5′-end of the sgRNA can be truncated to contain 17–18 nucleotides (Fig. 2b), hence increasing the sensitivity of sgRNA to mismatches without sacrificing on-target editing efficiency [47, 48]. On the contrary, truncation at the 3′-end of the sgRNA or by more than 3 nucleotides (16-nucleotide sgRNA) results in loss of on-target cleavage [18, 48, 49]. Truncating the 5′-end of the sgRNA is thought to increase the binding energy requirement between the sgRNA and DNA sequence, thus lowering the affinity of the sgRNA to off-target loci [48]. Inclusion of 2 guanine nucleotides at the 5′ end of the sgRNA also lowered the rate of OTEs significantly (Fig. 2c) but editing at on-target sites could also be reduce depending on the guide sequence [25, 50]. The underlying mechanism behind this change in specificity is unknown and intriguing given that an increase in GC content usually stabilises the hybridisation of RNA to DNA [51]. Finally, chemically modifying the central region of the 20-nucleotide guide sequence of the sgRNA through the inclusion of bridged or locked nucleic acids minimised OTEs [52]. The bridged/locked nucleic acids are thought to disrupt the stable state of the hybridised sgRNA-DNA complex at off-target sites, hence impeding the formation of DSBs.
Optimising the specificity of the Cas9 endonuclease
Cas9 protein from S. pyogenes contains the HNH and RuvC nuclease domains that cleave the DNA strands complementary and non-complementary to the guide sequence in sgRNA respectively [5]. Mutating either one of the catalytic residues of the nuclease domains (H840A in HNH and D10A in RuvC) transforms the Cas9 nuclease into a nickase that could only generate single-strand breaks (SSBs) with an overhang instead of blunt end cuts by wild-type Cas9 [5, 6]. It has been shown that using D10A-mutated Cas9 with a pair of sgRNA targeting sequences in proximity on opposite DNA strands (paired nickases, Fig. 2d) reduced OTEs by 100- to 1500-fold [50, 53,54,55,56]. Two SSBs, one on each DNA strand, generated in proximity to each other, essentially create a composite DSB. The decrease in OTEs is in part due to the low probability for off-target DSBs in this context [57, 58] while off-target SSBs are also repaired efficiently by local enzymes [59]. Cas9 with an inactivated RuvC domain has also been found to be more efficient than one with an inactivated HNH domain [6, 53].
Additionally, fusing the DNA catalytic domain of the FokI endonuclease to a catalytically deactivated Cas9 protein (dCas9) also considerably reduces the OTEs of CRISPR/Cas9 gene editing [60,61,62]. The dCas9 protein serves as the DNA recognition domain while the FokI nuclease domain induces a DSB following FokI dimerisation at the correct spacing and orientation (Fig. 2e). Since the FokI-dCas9 fusion protein requires a stricter target spacing of 15–25 base pairs for FokI dimerisation than for paired Cas9 nickases, the resulting specificity of the FokI-dCas9 protein is higher [60]. Despite the obligatory dimerisation of FokI nucleases to generate a DSB, indels, most likely arising from FokI monomers recruiting other monomers from solution, could still be detected at a lower frequency [62]. However, off-target indels induced by FokI monomers can be reduced with the use of truncated sgRNAs. The enhanced specificity of the paired Cas9 nickases and FokI-dCas9 protein is not without its drawbacks—the increased payload of the CRISPR/Cas9 components hinders effective delivery to the target cells/tissues.
Recent developments in CRISPR/Cas9 technology have also seen the emergence of base editors which can convert a single base into another without having to induce a DSB. A Cas9 nickase with a non-catalytic RuvC domain fused to APOBEC1, a cytidine deaminase enzyme, and uracil DNA glycosylase inhibitor (UGI) successfully converted a cytosine base to thymine (Fig. 2f) at the target site without having to create a DSB [63]. Similarly, the adenine base editor consisting of tRNA adenine deaminase and Cas9 nickase converts an adenine base to guanine [64]. These studies suggest the possibility of treating genetic disorders of point mutation origins at a lower risk of incurring OTEs. However, base editors can be limited by the narrow 5-nucleotide deamination window at the PAM-distal end of the protospacer as well as its inability to discriminate between specific cytosine residues within that window [63, 65]. In vitro assays using Digenome-seq and EndoV-seq have revealed higher genome-wide specificities of cytidine (9-fold) and adenine (2–20 fold) base editors when compared to Cas9 nuclease editing [66,67,68]. Recent studies adopting a more sensitive genome-wide detection of single-nucleotide variations confirmed that the cytidine base editors induced a 20-fold increase in single-nucleotide variations in mouse embryo when compared to controls while adenine base editors generated minimal single-nucleotide variations [69]. Since the nucleotide variations were predominantly cytosine to thymine conversions, these results further suggested that the cytidine base editor was potentially more promiscuous in on-target selection than the adenine base editor [69].
One of the prevailing theories hypothesise that specificity of the Cas9 endonuclease is governed by the binding energy threshold between the sgRNA-Cas9 complex and the DNA sequence. Wild-type sgRNA-Cas9 complexes contain more than optimal levels of energy for the binding of the complex to the target DNA locus; hence, the excess energy is able to accommodate off-target binding [48]. Through mutating the residues in the Cas9 protein which form hydrogen bonds with the DNA phosphate backbone (high-fidelity SpCas9-HF1) or substituting positively-charged amino acids with neutral alanine residues (enhanced specificity eSpCas9(1.1)), the Cas9 affinity for non-targeted sequences is diminished and the resulting Cas9 variants showed reduced OTEs [70, 71].
Recent studies on SpCas9-HF1 and eSpCas9(1.1) DNA binding assays, however, dispute the abovementioned hypothesis, showing that the binding affinities for on-target and off-target DNA sequences are comparable to wild-type SpCas9 [72]. Using single-molecule Förster resonance energy transfer (smFRET) techniques, the authors revealed that the HNH domains of SpCas9-HF1 and eSpCas9(1.1) remained in a catalytically inactive state when the nucleases were bound to off-targets [72]. In this way, alanine substitutions in these high-fidelity Cas9 proteins increase the threshold required for the HNH domain to switch to a conformationally active state [41, 73]. Based on these observations, a new hyper-accurate SpCas9 variant (HypaCas9) was designed to contain alanine substitutions in the non-catalytic REC3 domain, which restricted a downstream conformation change to an activated HNH domain when Cas9 is bound to off-targets [72]. When compared to SpCas9-HF1 and eSpCas9(1.1), HypaCas9 exhibited even greater specificity than the other 2 variants. GUIDE-seq assay revealed that with the sgRNA targeting VEGFA site 2, 18 off-target sites were detected for HypaCas9 while 19 and 24 off-target sites were detected for eSpCas9(1.1) and SpCas9-HF1, respectively [72]. Other high-fidelity variants of SpCas9 nucleases such as evoCas9 and xCas9 were developed through library screens of REC3-mutated SpCas9 and phage-assisted continuous evolution respectively [74, 75]. Similar to SpCas9-HF1, eSpCas9(1.1) and HypaCas9, evoCas9 and xCas9 displayed substantial reductions in off-target editing compared to wild-type SpCas9 while having near wild-type on-target efficiency. Under the GUIDE-seq analysis, evoCas9 showed a 98.7% reduction in genome-wide off-target indels compared to wild-type SpCas9 while at known promiscuous sites such as HEK site 4 and VEGFA, xCas9 showed a 4.2- to 9.4-fold reduction in off-target/on-target ratios compared to wild-type SpCas9 [74, 75].
One of the requirements for a Cas9-induced DSB is the presence of proximal PAM sites to the targeted DNA sequences [5]. The precision of the cleave site may therefore be restricted by the location of PAMs within the vicinity of the targeted sequence. To circumvent this requirement, several measures can be adopted including (1) engineering SpCas9 to recognise alternative PAM sequences, (2) Cas9 orthologs from other bacteria such as Staphylococcus aureus (SaCas9), and (3) other types of CRISPR/Cas system such as the type V Cas12a (formerly Cpf1) that possess different PAM preferences can be used.
In one study, residues in the PAM-interacting domain of SpCas9 that contact the nucleotides of the PAM sequence were mutated under different permutations and through a bacterial selection system, the resulting SpCas9 variants were segregated based on their ability to discriminate between the canonical 5′-NGG and non-canonical 5′-NGA PAM [76]. Previous reports have documented that wild-type SpCas9 is able to induce DSBs at non-canonical PAMs at a lower frequency [46, 53]. One of the variants with a D1135E mutation were able to demonstrate a greater discernment against non-canonical PAMs when tested in human cell lines while maintaining comparable activity at targeted sequences with canonical PAMs to wild-type SpCas9 [76]. The augmented specificity of the D1135E mutant also extended to off-target sites that do not contain non-canonical PAMs. In the same study, 2 SpCas9 variants, VRQR and VRER, with mutations at positions 1135, 1218, 1335 and 1337, were found to possess an enhanced affinity for 5′-NGA and 5′-NGCG PAM sites over wild-type SpCas9, respectively. The availability of SpCas9 variants with an expanded range of PAM preferences could therefore grant the accessibility to previously hard-to-target genes as well as limit the frequency of OTEs owing to the less commonly occurring PAMs [75].
Alternatively, Cas9 orthologs from other species such as SaCas9 (Fig. 2g) with a longer PAM requirement of 5′-NNGRRT (R = A or G) can be used to mediate gene editing in vivo with a similar efficiency and specificity to SpCas9 [76, 77]. The advantage of a smaller-sized SaCas9 protein relieves the issue of packaging into viral delivery systems with a limited payload and the requirement for a less frequently occurring PAM innately lowers the probability of OTEs [77]. The SaCas9 protein can also be modified to accept laxer PAM sequences such as the KKH-SaCas9 variant with a 5′-NNNRRT PAM requirement; however, this can translate to an increased likelihood of OTEs compared to native SaCas9 [78].
Finally, the Cas12a nuclease from Acidaminococcus sp. BV3L6 and Lachnospiraceae bacterium ND2006 (Fig. 2h) offers an attractive and higher fidelity option to CRISPR/Cas9-mediated genome editing of human cell lines [79, 80]. In comparison to CRISPR/Cas9 systems, the type V CRISPR/Cas12a system is able to process pre-crRNA into mature crRNA without a tracrRNA, hence reducing the size of the plasmid constructs [79]. In a recent study, a single, carefully designed pre-crRNA had been shown to be sufficient in driving simultaneous multiplex gene editing in HEK293T cells and mouse neurons in vivo [81]. The Cas12a protein recognises a T-rich (5′-TTTN) PAM sequence instead of the 5′-NGG PAM by its Cas9 counterpart, thus offering a greater precision in targeting novel gene loci [79]. This has been reflected in a study that utilised the Cas12a nuclease to correct mutations in the dystrophin gene (DMD) that causes Duchenne muscular dystrophy (DMD) [82]. Additionally, Cas12a-induced DSBs generate a 5′ overhang of 4–5 nucleotides that can facilitate gene insertion via the NHEJ repair pathway as opposed to the blunt end cuts generated by Cas9 [5, 79, 83]. Like the SpCas9 and SaCas9 endonucleases, Cas12a can be engineered to improve on-target activity and reduce off-target mutations in human cell line editing [84].
In summary, Table 1 provides a consolidated overview of the aforementioned engineered Cas9 variants, Cas9 orthologs and Cas nuclease from other systems.
Modality of the CRISPR/Cas9 system during delivery
The CRISPR/Cas9 system consisting of the sgRNA and Cas9 nuclease can be delivered in several forms into the host cell for genome editing. The cDNA of the Cas9 nuclease as well as the sgRNA can be expressed in a single plasmid [44] or separated into 2 plasmids to be delivered concurrently into the host cell [7]. While the design and cloning of the expression cassette is relatively non-complicated, the use of plasmid DNA can potentially lead to the integration of unwanted DNA sequences into the host genome [87]. Prolonged expression of the sgRNA and Cas9 nuclease due to the persistence of plasmid DNA can also increase the potential of OTEs [88]. In addition, introduction of foreign DNA can trigger cellular immune responses [89]. One study proposed an engineered plasmid expression cassette to contain 2 sgRNAs, one targeting the gene of interest and the other targeting the Cas9 itself [90]. Designed in this manner, the Cas9 transgene would be cleaved simultaneously with the gene of interest, hence reducing the duration of Cas9 expression. As expected, the Cas9 nuclease expression level was found to be diminished by post-treatment day 2 while measured OTEs were significantly reduced correspondingly [90].
The Cas9 endonuclease can be delivered in the form of mRNA into the host cell. Switching to Cas9 mRNA prevents the integration of plasmid DNA into the host genome and reduces the propensity for OTEs by reducing the exposure time to Cas9 nuclease [91, 92]. Delivering the Cas9 nuclease as a mRNA precursor also hastens the onset of CRISPR/Cas9-mediated gene editing since the process bypasses the transcription of Cas9 cDNA [91].
Finally, Cas9 can be delivered as a protein complexed with the mature sgRNA to form a ribonucleoprotein (RNP) assembly [91, 93,94,95]. Cas9 RNPs are able to generate genomic on-target DSBs immediately after delivery and are rapidly broken down by endogenous proteases [91, 96]. By titrating the concentration of the Cas9 RNPs to an optimal level and limiting the window of exposure to the RNPs, OTEs in the edited cells can be substantially reduced by 2.2 to 19 folds [91, 93, 95, 96]. Similar to using Cas9 mRNA, cellular toxicity associated with exogenous DNA and the possibility of foreign DNA integration can be avoided with Cas9 RNP [97].
DNA repair pathways
Resolution of DSBs generated by CRISPR/Cas9-mediated gene editing can proceed via 2 pathways: NHEJ and homology-directed repair (HDR). In mammalian cells, NHEJ is the predominant repair pathway that competes with the less efficient HDR pathway [98, 99]. NHEJ is an error-prone but efficient pathway that creates indels, leading to frameshift mutations and eventually functional gene knockouts [8, 100]. Using 2 DSBs simultaneously, the exon-coding sequence or regulatory element of genes can also be deleted and the DNA cleavage rectified via NHEJ [101,102,103]. In tumourigenesis, where mutated oncogenes such as KRAS and EGFR initiate the proliferation and survival of cancerous cells, the CRISPR/Cas9 system can generate DSBs within the exon-coding regions of the mutated alleles, which is then repaired via NHEJ to form indels that disrupt the oncogenes and inhibit the survival and tumourigenicity of the mutant cells [104, 105]. This CRISPR/Cas9-mediated anti-tumourigenic effect has also been replicated in in vivo xenograft models of cancer, with additional beneficial effects of abrogating angiogenesis and metastasis of the cancerous cells [105,106,107,108].
However, gene therapy can also require the correction of a defective gene through the repair of a point mutation or the precise integration of a functional gene copy into the genome of the target cells. This can be accomplished through the HDR pathway with an exogenously supplied donor repair template, often in the form of single-stranded oligodeoxynucleotides (ssODNs) for point mutation corrections or plasmid DNA templates for entire transgenes [6, 7, 109, 110]. Successful HDR-mediated gene editing is determined by the length of the repair template and flanking sequences that are homologous to either side of the DSB. Larger repair templates require longer homology arms with a minimum of 400 base pairs for efficient editing [111, 112]. In contrast, ssODNs do not require long homology arms to correct point mutations [100]. Improvement in HDR efficiency can also be achieved through using asymmetrical donor template [113, 114].
To bias the DNA repair mechanisms towards HDR-mediated pathways, studies have reported using small-molecule inhibitors such as Scr7, shRNA and proteins that target DNA ligase IV, an important enzyme in the NHEJ pathway [99, 115] and RS-1, an HDR enhancer that exerts its effect through stimulating the human HR RAD51 protein [116]. Others have also synchronised the delivery of the sgRNA and Cas9 nuclease to the late G2 phase of the cell cycle or fusing the Cas9 nuclease to the human Geminin protein to capture the active time window of HDR pathways during late S and G2 phases of the cell cycle [117, 118]. Additionally, the donor template can be tethered to the Cas9 nuclease to improve HDR efficiency [119, 120].
However, despite these innovative executions to favour the HDR repair pathway, precise gene editing remains a challenge in post-mitotic cell populations such as neurons since HDR is confined to the late S and G2 phases of the cell cycle. The emergence of base editors thus presents an attractive alternative for inducing single nucleotide mutation in these cell populations since the repair mechanism does not depend on HR pathways [63, 64, 121]. Likewise, other studies have hijacked the NHEJ repair machinery in developing a homology-independent targeted integration (HITI) which demonstrated more robust knock-in efficiencies for the MERTK gene in postnatal mouse neurons as compared to the traditional HDR method [122]. The transgene is integrated in a specified orientation since the sgRNA/SpCas9 complex is designed to cleave off the donor DNA if it is inserted in the reverse direction [123].
Targeting specific tissue/cell populations in vivo
In most mammalian systems, there is a very low likelihood of a homogenous cellular composition within any given tissue or organ. This presents a considerable barrier when genome editing is to be restricted to a selected population of cells in the tissue. The expressions of Cas9 endonuclease and sgRNA are typically driven by RNA polymerase II and III promoters respectively [124]. Viral RNA polymerase II promoters such as cytomegalovirus (CMV) and simian virus 40 (SV40) and eukaryotic promoters such human elongation factor 1 alpha (EF1α) and chicken β-actin (CBA) RNA polymerase II promoters are commonly used and are constitutively active [125, 126]. Viral RNA polymerase II promoters are preferred over eukaryotic promoters because viral promoters induce a higher level of transcription [127]. Expression of the sgRNA is usually regulated by an RNA polymerase III promoter such as the human U6 promoter which transcribes genes that encode for small RNA sequences in eukaryotes [124, 128]. This is because sgRNA expressed under the RNA polymerase II promoter is non-functional when unique sequences like the 5′-cap and 3′-polyadenylation tail are included in the transcript [124].
Choice of promoter regions and inducible CRISPR/Cas9 systems
Since most of the frequently-used RNA polymerase II and III promoters in CRISPR/Cas9 genome editing are ubiquitously expressed in many cell types, they are not feasible when cell-type specific editing is required [125]. Hence, switching over to tissue-specific promoters to drive the targeted expression of Cas9 endonuclease should be considered when designing the vector plasmid. For example, the promoters for hsynapsin and cardiac troponin T (cTnT) are respective neuron- and cardiomyocyte-specific RNA polymerase II promoters that drive gene expression downstream [129,130,131]. A selected list of cell-specific promoters is available in Table 2. On a precautionary note, many native promoters, once thought to dictate gene expression in a specific cell type, are also active in other cell populations. Glial fibrillary acidic protein (GFAP) is predominantly expressed in astrocytes [132], but it can also be found in human keratinocytes and testicular Leydig cells [133, 134].
The integration of an inducible promoter upstream of the tissue-specific promoter can impose spatial and temporal control over Cas9 expression, culminating in lower OTE frequencies given that one of the determinants of OTE rates is the length of time the genome is exposed to the nuclease [47, 91]. Studies have used the TRE3G (Tet-On-3G) and TRE2 Tet-On promoters to activate Cas9 proteins in an animal model and human immortalised cell lines respectively under the control of administered doxycycline [146, 147]. Lower OTEs were observed when compared to constitutively active Cas9 controls and embryonic lethality was avoided in the mouse model when the APC and TRP53 tumour suppressing genes were targeted only during young adulthood [146]. This design could be adapted to regulate sgRNA expression and to restrict CRISPR/Cas9 editing to selected cell types in vivo within a designated time period or developmental stage [148, 149].
Other inducible systems aimed at regulating the sgRNA and Cas9 time window of activity have been described in previous literature. These include the use of ligand-activated aptazyme-embedded guide RNA [150], allosteric modulation of ligand-sensitive Cas9 [151], and conditionally-activated Cas9-intein and Cas9-destabilised domain fusion proteins [152,153,154]. Alternatively, temperature-sensitive Cas9 variants that operate within clinically tolerable ranges can be considered as a viable inducible option that avoids the potential toxicity of small-molecule ligands [155].
microRNAs and anti-CRISPR proteins
More recently, studies have harnessed the cell-type specificity of microRNAs (miRNA) and anti-CRISPR (Acr) proteins in refining the fidelity of CRISPR/Cas9-mediated editing [156]. Acr proteins are derived from prophages that evolve to escape bacterial immunity by inhibiting the CRISPR/Cas defence mechanism [157]. Among the Acr proteins, AcrIIA2 and AcrIIA4 isolated from the Listeria monocytogenes prophage specifically target the SpCas9 nuclease, with AcrIIA4 in particular interfering with DNA recognition at the PAM-interacting domain and hindering the RuvC catalytic domain [157,158,159,160]. On the other hand, miRNAs are short, non-coding RNA sequences that regulate protein expression by targeting their mRNA for degradation or inhibiting the translation pathway [161].
Taking advantage of both systems, miRNA response elements (MREs) can be inserted into the 5′- or 3′-UTR of AcrIIA4 transgenes so that Acr protein expression levels can be regulated by cell-specific miRNAs [162,163,164]. Using a cardiomyocyte-specific miR-1, the expression of AcrIIA4 is repressed, allowing the sgRNA-SpCas9 complex to bind and cleave the target DNA sequence within the genome of cardiomyocytes. Conversely, since miR-1 is not present in off-target cells, AcrIIA4 is highly expressed and thereby inhibits sgRNA-SpCas9 binding to DNA [164]. This technique was replicated with miR-122 which demonstrated specificity in hepatocytes.
Cell-type-specific miRNA-mediated genome editing can also be achieved through a simplified protocol. In one study, Wang and colleagues designed a unique pre-sgRNA construct that consisted of an sgRNA flanked by an MRE on each side under the influence of the CAGGS RNA polymerase II promoter [165]. Due to the presence of a 5′-cap and 3′-polyadenylation tail, the pre-sgRNA remained non-functional until the regulatory elements were cleaved off by endo- or exogenous cell-type specific miRNA. CRISPR/Cas9-mediated genome editing can therefore be restricted to targeted cell types that express the complementary miRNAs to the MREs [165].
Despite the improved specificity conferred by the inducible and the Acr/miRNA-mediated CRISPR/Cas9 gene editing systems, several issues have to be addressed before these techniques can be translatable clinically. Small-molecule ligands can induce cytotoxicity and immunogenic responses [166] while the leakiness of Tet-On/Tet-Off systems limits its use in patient populations [146, 148]. The delivery efficiency of a large payload that includes the Acr and Cas9 transgenes, sgRNA and the repair template to the same cell may be low, while the persistence of foreign Acr proteins beyond a few days raises significant concerns.
Targeted delivery of the CRISPR/Cas9 system in vivo
In vivo gene delivery approaches can be broadly divided into viral and synthetic non-viral vectors that are either locally or systemically administered. Viral vectors are able to mediate efficient gene transfer to the target cells but they carry the potential to elicit immunogenic and cytotoxic responses [167,168,169]. Moreover, packaging the CRISPR/Cas9 components into the virus can be a challenge. The cargo size limit for the popular adeno-associated virus (AAV) vector is around 4.7 k bp [170] while the size of the commonly-used SpCas9 is around 4.2 k bp alone [171]. While it is possible to concomitantly deliver SpCas9 and the sgRNA in the same vector, there is little room available for the inclusion of regulatory elements and donor repair templates [171, 172]. To bypass this issue, SpCas9 can be truncated [173] or split into 2 domains delivered separately [174]. SpCas9 can also be substituted by smaller Cas9 orthologs such as the ~3.2 k bp SaCas9 [77, 85].
In comparison, non-viral synthetic vectors carry a lower risk of initiating immunogenic events and lack the viral machinery to integrate exogenous DNA material into the host genome [175]. The payload capacity can also be expanded to contain the sgRNA, Cas9 nuclease and donor template within a single vector with ease [176] or to deliver the CRISPR/Cas9 components as an RNP [95, 177]. Furthermore, synthetic vectors are easy to manufacture in a large scale [178]. However, the main drawback of a non-viral vector system is that the delivery efficiency is often several folds lower than viral-mediated gene delivery [175, 179]. Despite the growing interest in synthetic vectors, the majority of the clinical trials involving gene therapy still use locally administered viral vectors because of the more robust delivery efficiency while restricting systemic off-targets [180].
Non-viral delivery of CRISPR/Cas9 components encapsulates the contents within a lipid, polymer or inorganic carrier or by conjugating the sgRNA/Cas9 nuclease with peptide sequences. Using a synthetic vector offers the option of delivering the sgRNA and Cas9 nuclease as an RNP which reduces the probability of OTEs by lowering the exposure time to the sgRNA/Cas9 nuclease [91, 95, 176]. Furthermore, synthetic vectors can be engineered to target specific cell populations in vivo by incorporating surface ligands onto vectors that would recognise and bind to distinct receptors on the target [181, 182]. These ligands can take the form of an organic molecule, antibody, aptamer or protein/peptide and permit the vectors to differentiate between healthy tissues and tumour cells [181, 182]. A recent study demonstrated that conjugation of folic acid molecules to polyethylene glycol-succinyl-Chol liposomes facilitated the targeting of the CRISPR/Cas9 vector to ovarian carcinomas where surface folate receptors are expressed in abundance [183, 184]. The increased proximity between the vector and target cell as a consequence of folic acid ligand-folate receptor binding initiates the internalisation of the vector via endocytosis, followed by the release of the vector contents into the cell cytoplasm [184]. In a similar fashion, transferrin ligands can be inserted into the surface of liposomes to target ovarian cancer cells, which also express transferrin receptors in high levels [185]. Additionally, these techniques can be modified to integrate antibodies and peptides such as Angiopep-2 into the synthetic vectors, enabling blood-brain barrier permeation and subsequent gene editing of glioblastoma-associated cells [186,187,188]. Lastly, cell-based systematic evolution of ligands by exponential enrichment has also generated novel cell-type specific aptamers (single-stranded DNA or RNA oligonucleotides) that could substitute as cell recognition moieties on the vectors targeting osteosarcomeric cells in vivo [189].
Off-target effects of CRISRP/Cas9 editing in clinically relevant animal models
The repository website, https://clinicaltrials.gov, currently (retrieved August 2, 2019) lists a total of 28 clinical trials where the use of CRISPR/Cas9 technology has been approved in patient treatment. Of the 28 listed trials, 5 have been suspended or withdrawn. In one clinical trial, HIV-positive patients who had developed AIDS and haematological malignancies were infused with allogeneic CD34+ haematopoietic stem/progenitor cells that had the CCR5 gene ablated by the CRISPR/Cas9 tool to attenuate disease progression [190]. While data from the clinical trial is currently not available, studies have been conducted whereby human CD34+ haematopoietic stem/progenitor cells with CCR5 gene disruption were infused into immunodeficient mice [191]. The CCR5 editing efficiency was established to be at around 30%, with a detectable population of edited stem/progenitor cells after 30–47 weeks. These long surviving stem/progenitor cells were able to self-renew and differentiate into multiple cell types of haematopoietic lineage. When exposed to the HIV-1 virus, the mice infused with stem/progenitor cells containing the mutated CCR5 gene exhibited resistance to HIV-1, as evidenced by the decrease in HIV-1 RNA levels [191]. Careful design of the sgRNA template also minimised OTEs, with whole genome sequencing showing no indels at the closely homologous CCR2 gene locus and one potential off-target site placed within a nonsense region [191]. While the results were encouraging, detection of OTEs via whole genome sequencing are often limited by the cost associated with high sequencing depth [192, 193]. Hence, it is likely that low frequency OTEs may be missed when the sequencing depth is insufficient (< 10-fold) [194]. To optimise the balance between cost effectiveness and the sensitivity of genome-wide OTEs detection in vivo, a recent study has proposed a new strategy termed “Verification of In Vivo Off-targets” (VIVO) [195]. Briefly, VIVO consists of 2 stages: an in vitro and an in vivo stage. The in vitro stage identifies potential off-target cleavages by CRISPR/Cas9 treatment in vitro via CIRCLE-seq, a next-generation sequencing technique with higher sensitivity for OTEs (due to lower background) than contemporary cell-based detection approaches [196]. The second stage involves the confirmation of off-target sites identified by CIRCLE-seq. Targeted amplicon sequencing was carried out on off-target sites selected for their CIRCLE-seq read counts, in liver tissue harvested from mice treated with viral vectors containing CRISPR/Cas9 components [195]. When the sgRNA targeting the mouse PCSK9 gene was switched from a less discriminating design to that which were aligned more orthogonally with the mouse genome, no off-target indels (excluding the human PCSK9 transgene) could be detected by VIVO [195]. The study validated the robustness and sensitivity of VIVO to off-target indels generated by in vivo CRISPR/Cas9 edits, hence presenting a strong claim for its application in clinical therapy [195].
Chimeric antigen receptor (CAR) T cells are T cells genetically engineered to express CARs that contain antigen-recognition and T cell activating domains [197]. CARs facilitate T cell targeting of tumours by binding to specific antigens present on the cancer cell surface and activating cytotoxic pathways to eliminate the cancer cells [198]. In a recently approved clinical trial [199], T cells were transduced with gammaretroviral vectors carrying a CD7 CAR and CD28 endodomain to treat T cell leukaemia/lymphoma [200]. Since T cell lymphomas and non-malignant T cells both express the CD7 glycoprotein, CRISPR/Cas9 gene editing was performed before viral transduction to disrupt the endogenous CD7 gene and to avoid self-targeting in the CAR T-cells [199]. In vitro studies showed that CD7 CAR T-cells reduced malignant CD7-positive cell lines by at least 95% in cocultures while no observable cytotoxic effect was reported in cocultures with CD7-negative cell lines [200]. Infusing the CD7 CAR T-cells into immunodeficient mice engrafted with tumourigenic CCRF-CEM cells halted the development of leukaemia and prolonged the survival period of the mice. Whole genome sequencing of the CD7 CAR T-cell following CRISPR/Cas9-mediated disruption of the CD7 gene revealed no significant OTEs when compared to sham controls [200].
Gleaning from the aforementioned complementary preclinical studies, factors such as well-designed, truncated sgRNAs [191] and the delivery of sgRNAs and Cas9 nuclease as an RNP [200] can abate the incidences of OTEs. Furthermore, there are many other in vivo preclinical disease models where rare or non-occurrence of OTEs following CRISPR/Cas9 genome editing lends credibility to the high specificity achievable by this genome editing tool [201,202,203,204,205,206,207].
Conclusion
A well-designed CRISPR/Cas9 study or clinical trial takes into consideration the different aspects in which the gene editing tool can be fully optimised to attain maximal on-target efficiency and minimising OTEs. The absence or rare occurrence of OTEs in preclinical/humanised animal models and clinical pilot studies offers proof of principle that the current level of specificity in CRISPR/Cas9 genome editing permits this technique to be translated onto a larger clinical scale. From a different perspective, the clinician may be forced to choose between administering the gene therapy and incurring the risk of off-target mutations or to forgo therapy and miss the opportunity to diffuse a life-threatening condition. Moreover, if the off-target mutations are non-lethal, will the patient benefit more from gene therapy while coping with the mutation-induced side effects through pharmacological interventions? Lastly, the broad applicability of the CRISPR/Cas9 editing technique demands for strong regulatory institutions and medical ethics boards to prevent any abuse or ethical/moral transgressions, as in the case of Jesse Gelsinger [208] and the CCR5 gene-edited Chinese babies [209]. Ultimately, the country’s judicial and regulatory bodies have to take into account the political, societal and cultural ideologies and weigh the benefits and risks of gene therapy on a case-by-case basis.
Abbreviations
- ADA−SCID:
-
ADA-linked severe combined immuno-deficiency
- HR:
-
Homologous recombination
- ZFN:
-
Zinc-finger nuclease
- TALEN:
-
Transcription activator-like effector nuclease
- CRISPR:
-
Clustered regularly interspaced short palindromic repeats
- DSB:
-
Double-strand break
- NHEJ:
-
Non-homologous end joining
- OTE:
-
Off-target effect
- pre-crRNA:
-
Precursor CRISPR RNA
- tracrRNA:
-
Trans-activating crRNA
- PAM:
-
Protospacer adjacent motif
- SpCas9:
-
Streptococcus pyogenes Cas9
- sgRNA:
-
Single-guide RNA
- SSB:
-
Single-strand break
- dCas9:
-
Deactivated Cas9
- indel:
-
Insertion/deletion
- UGI:
-
Uracil DNA glycosylase inhibitor
- smFRET:
-
Single-molecule Förster resonance energy transfer
- SaCas9:
-
Staphylococcus aureus Cas9
- DMD:
-
Duchenne muscular dystrophy
- RNP:
-
Ribonucleoprotein
- CMV:
-
Cytomegalovirus
- SV40:
-
Simian virus 40
- EF1α:
-
Elongation factor 1 alpha
- CBA:
-
Chicken β-actin
- cTnT:
-
Cardiac troponin T
- GFAP:
-
Glial fibrillary acidic protein
- miRNA:
-
MicroRNA
- Acr:
-
Anti-CRISPR
- MRE:
-
miRNA response element
- VIVO:
-
Verification of in vivo off-targets
- CAR:
-
Chimeric antigen receptor
References
Blaese RM, Culver KW, Miller AD et al (1995) T lymphocyte-directed gene therapy for ADA- SCID: initial trial results after 4 years. Science 270:475–480
Bibikova M, Beumer K, Trautman JK, Carroll D (2003) Enhancing gene targeting with designed zinc finger nucleases. Science 300:764
Miller JC, Tan S, Qiao G et al (2011) A TALE nuclease architecture for efficient genome editing. Nat Biotechnol 29:143–148
Ho BX, Loh SJH, Chan WK, Soh BS (2018) In vivo genome editing as a therapeutic approach. Int J Mol Sci 19:2721
Jinek M, Chylinski K, Fonfara I et al (2012) A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337:816–821
Cong L, Ran FA, Cox D et al (2013) Multiplex genome engineering using CRISPR/Cas systems. Science 339:819–823
Mali P, Yang L, Esvelt KM et al (2013) RNA-guided human genome engineering via Cas9. Science 339:823–826
Gaj T, Gersbach CA, Barbas CF (2013) ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol 31:397–405
Tebas P, Stein D, Tang WW et al (2014) Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N Engl J Med 370:901–910
Qasim W, Zhan H, Samarasinghe S et al (2017) Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR T cells. Sci Transl med 9:eaaj2013
Vertex Pharmaceuticals Incorporated, CRISPR Therapeutics A Safety and Efficacy Study Evaluating CTX001 in Subjects with Transfusion-Dependent β-Thalassemia. In: NLM Identifier: NCT03655678. https://clinicaltrials.gov/show/NCT03655678. Accessed 8 Aug 2019
University of Pennsylvania, Parker Institute for Cancer Immunotherapy, Tmunity Therapeutics NY-ESO-1-redirected CRISPR (TCRendo and PD1) Edited T Cells (NYCE T Cells). In: NLM Identifier: NCT03399448. https://clinicaltrials.gov/show/NCT03399448. Accessed 8 Aug 2019
Gabriel R, Lombardo A, Arens A et al (2011) An unbiased genome-wide analysis of zinc-finger nuclease specificity. Nat Biotechnol 29:816–823
Li H, Haurigot V, Doyon Y et al (2011) In vivo genome editing restores haemostasis in a mouse model of haemophilia. Nature 475:217–221
Pattanayak V, Ramirez CL, Joung JK, Liu DR (2011) Revealing off-target cleavage specificities of zinc-finger nucleases by in vitro selection. Nat Methods 8:765–770
Anguela XM, Sharma R, Doyon Y et al (2013) Robust ZFN-mediated genome editing in adult hemophilic mice. Blood 122:3283–3287
Poirot L, Philip B, Schiffer-Mannioui C et al (2015) Multiplex genome-edited T-cell manufacturing platform for “off-the-shelf” adoptive T-cell immunotherapies. Cancer Res 75:3853–3864
Fu Y, Foden JA, Khayter C et al (2013) High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotechnol 31:822–826
Duan J, Lu G, Xie Z et al (2014) Genome-wide identification of CRISPR/Cas9 off-targets in human genome. Cell Res 24:1009–1012
Kuscu C, Arslan S, Singh R et al (2014) Genome-wide analysis reveals characteristics of off-target sites bound by the Cas9 endonuclease. Nat Biotechnol 32:677–683
Lin Y, Cradick TJ, Brown MT et al (2014) CRISPR/Cas9 systems have off-target activity with insertions or deletions between target DNA and guide RNA sequences. Nucleic Acids Res 42:7473–7485
Wang X, Wang Y, Wu X et al (2015) Unbiased detection of off-target cleavage by CRISPR-Cas9 and TALENs using integrase-defective lentiviral vectors. Nat Biotechnol 33:175–178
Liang P, Xu Y, Zhang X et al (2015) CRISPR/Cas9-mediated gene editing in human tripronuclear zygotes. Protein Cell 6:363–372
Hsu PD, Lander ES, Zhang F (2014) Development and applications of CRISPR-Cas9 for genome engineering. Cell 157:1262–1278
Kim D, Bae S, Park J et al (2015) Digenome-seq: genome-wide profiling of CRISPR-Cas9 off-target effects in human cells. Nat methods 12:237–243 1 p following 243
Doench JG, Fusi N, Sullender M et al (2016) Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat Biotechnol 34:184–191
Listgarten J, Weinstein M, Kleinstiver BP et al (2018) Prediction of off-target activities for the end-to-end design of CRISPR guide RNAs. Nat Biomed Eng 2:38–47
Bolotin A, Quinquis B, Sorokin A, Ehrlich SD (2005) Clustered regularly interspaced short palindrome repeats (CRISPRs) have spacers of extrachromosomal origin. Microbiology (Reading, Engl) 151:2551–2561
Mojica FJM, Díez-Villaseñor C, García-Martínez J, Soria E (2005) Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J Mol Evol 60:174–182
Pourcel C, Salvignol G, Vergnaud G (2005) CRISPR elements in Yersinia pestis acquire new repeats by preferential uptake of bacteriophage DNA, and provide additional tools for evolutionary studies. Microbiology (Reading, Engl) 151:653–663
Barrangou R, Fremaux C, Deveau H et al (2007) CRISPR provides acquired resistance against viruses in prokaryotes. Science 315:1709–1712
Marraffini LA (2015) CRISPR-Cas immunity in prokaryotes. Nature 526:55–61
Westra ER, Dowling AJ, Broniewski JM, van Houte S (2016) Evolution and ecology of CRISPR. Annu Rev Ecol Evol Syst 47:307–331
Wright AV, Nuñez JK, Doudna JA (2016) Biology and applications of CRISPR systems: harnessing nature’s toolbox for genome engineering. Cell 164:29–44
Sander JD, Joung JK (2014) CRISPR-Cas systems for editing, regulating and targeting genomes. Nat Biotechnol 32:347–355
Brouns SJJ, Jore MM, Lundgren M et al (2008) Small CRISPR RNAs guide antiviral defense in prokaryotes. Science 321:960–964
Deltcheva E, Chylinski K, Sharma CM et al (2011) CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 471:602–607
Lim Y, Bak SY, Sung K et al (2016) Structural roles of guide RNAs in the nuclease activity of Cas9 endonuclease. Nat Commun 7:13350
Sternberg SH, Redding S, Jinek M et al (2014) DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature 507:62–67
Mojica FJM, Díez-Villaseñor C, García-Martínez J, Almendros C (2009) Short motif sequences determine the targets of the prokaryotic CRISPR defence system. Microbiology (Reading, Engl) 155:733–740
Sternberg SH, LaFrance B, Kaplan M, Doudna JA (2015) Conformational control of DNA target cleavage by CRISPR-Cas9. Nature 527:110–113
Cui Y, Xu J, Cheng M et al (2018) Review of CRISPR/Cas9 sgRNA design tools. Interdiscip Sci 10:455–465
Cho SW, Kim S, Kim JM, Kim J-S (2013) Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat Biotechnol 31:230–232
Ran FA, Hsu PD, Wright J et al (2013) Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8:2281–2308
Moreno-Mateos MA, Vejnar CE, Beaudoin J-D et al (2015) CRISPRscan: designing highly efficient sgRNAs for CRISPR-Cas9 targeting in vivo. Nat Methods 12:982–988
Hsu PD, Scott DA, Weinstein JA et al (2013) DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol 31:827–832
Pattanayak V, Lin S, Guilinger JP et al (2013) High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat Biotechnol 31:839–843
Fu Y, Sander JD, Reyon D et al (2014) Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat Biotechnol 32:279–284
Hwang WY, Fu Y, Reyon D et al (2013) Heritable and precise zebrafish genome editing using a CRISPR-Cas system. PLoS One 8:e68708
Cho SW, Kim S, Kim Y et al (2014) Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res 24:132–141
Sugimoto N, Nakano S, Katoh M et al (1995) Thermodynamic parameters to predict stability of RNA/DNA hybrid duplexes. Biochemistry 34:11211–11216
Cromwell CR, Sung K, Park J et al (2018) Incorporation of bridged nucleic acids into CRISPR RNAs improves Cas9 endonuclease specificity. Nat Commun 9:1448
Mali P, Aach J, Stranges PB et al (2013) CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat Biotechnol 31:833–838
Ran FA, Hsu PD, Lin C-Y et al (2013) Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 154:1380–1389
Shen B, Zhang W, Zhang J et al (2014) Efficient genome modification by CRISPR-Cas9 nickase with minimal off-target effects. Nat Methods 11:399–402
Frock RL, Hu J, Meyers RM et al (2015) Genome-wide detection of DNA double-stranded breaks induced by engineered nucleases. Nat Biotechnol 33:179–186
Bolukbasi MF, Gupta A, Wolfe SA (2016) Creating and evaluating accurate CRISPR-Cas9 scalpels for genomic surgery. Nat Methods 13:41–50
Lee J, Chung J-H, Kim HM et al (2016) Designed nucleases for targeted genome editing. Plant Biotechnol J 14:448–462
Kim J-S (2016) Genome editing comes of age. Nat Protoc 11:1573–1578
Guilinger JP, Thompson DB, Liu DR (2014) Fusion of catalytically inactive Cas9 to FokI nuclease improves the specificity of genome modification. Nat Biotechnol 32:577–582
Tsai SQ, Wyvekens N, Khayter C et al (2014) Dimeric CRISPR RNA-guided FokI nucleases for highly specific genome editing. Nat Biotechnol 32:569–576
Wyvekens N, Topkar VV, Khayter C et al (2015) Dimeric CRISPR RNA-guided FokI-dCas9 nucleases directed by truncated gRNAs for highly specific genome editing. Hum Gene Ther 26:425–431
Komor AC, Kim YB, Packer MS et al (2016) Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533:420–424
Gaudelli NM, Komor AC, Rees HA et al (2017) Programmable base editing of a•T to G•C in genomic DNA without DNA cleavage. Nature 551:464–471
Kim YB, Komor AC, Levy JM et al (2017) Increasing the genome-targeting scope and precision of base editing with engineered Cas9-cytidine deaminase fusions. Nat Biotechnol 35:371–376
Kim D, Lim K, Kim S-T et al (2017) Genome-wide target specificities of CRISPR RNA-guided programmable deaminases. Nat Biotechnol 35:475–480
Kim D, Kim D, Lee G et al (2019) Genome-wide target specificity of CRISPR RNA-guided adenine base editors. Nat Biotechnol 37:430–435
Liang P, Xie X, Zhi S et al (2019) Genome-wide profiling of adenine base editor specificity by EndoV-seq. Nat Commun 10:67
Zuo E, Sun Y, Wei W et al (2019) Cytosine base editor generates substantial off-target single-nucleotide variants in mouse embryos. Science eaav9973. https://doi.org/10.1126/science.aav9973
Kleinstiver BP, Pattanayak V, Prew MS et al (2016) High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature 529:490–495
Slaymaker IM, Gao L, Zetsche B et al (2016) Rationally engineered Cas9 nucleases with improved specificity. Science 351:84–88
Chen JS, Dagdas YS, Kleinstiver BP et al (2017) Enhanced proofreading governs CRISPR-Cas9 targeting accuracy. Nature 550:407–410
Dagdas YS, Chen JS, Sternberg SH et al (2017) A conformational checkpoint between DNA binding and cleavage by CRISPR-Cas9. Sci Adv 3:eaao0027
Casini A, Olivieri M, Petris G et al (2018) A highly specific SpCas9 variant is identified by in vivo screening in yeast. Nat Biotechnol 36:265–271
Hu JH, Miller SM, Geurts MH et al (2018) Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature 556:57–63
Kleinstiver BP, Prew MS, Tsai SQ et al (2015) Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature 523:481–485
Ran FA, Cong L, Yan WX et al (2015) In vivo genome editing using Staphylococcus aureus Cas9. Nature 520:186–191
Kleinstiver BP, Prew MS, Tsai SQ et al (2015) Broadening the targeting range of Staphylococcus aureus CRISPR-Cas9 by modifying PAM recognition. Nat Biotechnol 33:1293–1298
Zetsche B, Gootenberg JS, Abudayyeh OO et al (2015) Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 163:759–771
Swarts DC, Jinek M (2018) Cas9 versus Cas12a/Cpf1: structure-function comparisons and implications for genome editing. Wiley Interdiscip rev RNA e1481. https://doi.org/10.1002/wrna.1481
Zetsche B, Heidenreich M, Mohanraju P et al (2017) Multiplex gene editing by CRISPR-Cpf1 using a single crRNA array. Nat Biotechnol 35:31–34
Zhang Y, Long C, Li H et al (2017) CRISPR-Cpf1 correction of muscular dystrophy mutations in human cardiomyocytes and mice. Sci Adv 3:e1602814
Maresca M, Lin VG, Guo N, Yang Y (2013) Obligate ligation-gated recombination (ObLiGaRe): custom-designed nuclease-mediated targeted integration through nonhomologous end joining. Genome Res 23:539–546
Kleinstiver BP, Sousa AA, Walton RT et al (2019) Engineered CRISPR-Cas12a variants with increased activities and improved targeting ranges for gene, epigenetic and base editing. Nat Biotechnol 37:276–282
Friedland AE, Baral R, Singhal P et al (2015) Characterization of Staphylococcus aureus Cas9: a smaller Cas9 for all-in-one adeno-associated virus delivery and paired nickase applications. Genome Biol 16:257
Kim D, Kim J, Hur JK et al (2016) Genome-wide analysis reveals specificities of Cpf1 endonucleases in human cells. Nat Biotechnol 34:863–868
Ono R, Yasuhiko Y, Aisaki K et al (2019) Exosome-mediated horizontal gene transfer occurs in double-strand break repair during genome editing. Commun Biol 2:57
Merkle FT, Neuhausser WM, Santos D et al (2015) Efficient CRISPR-Cas9-mediated generation of knockin human pluripotent stem cells lacking undesired mutations at the targeted locus. Cell Rep 11:875–883
Sun L, Wu J, Du F et al (2013) Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339:786–791
Chen Y, Liu X, Zhang Y et al (2016) A self-restricted CRISPR system to reduce off-target effects. Mol Ther 24:1508–1510
Kim S, Kim D, Cho SW et al (2014) Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res 24:1012–1019
Liang X, Potter J, Kumar S et al (2015) Rapid and highly efficient mammalian cell engineering via Cas9 protein transfection. J Biotechnol 208:44–53
Ramakrishna S, Kwaku Dad A-B, Beloor J et al (2014) Gene disruption by cell-penetrating peptide-mediated delivery of Cas9 protein and guide RNA. Genome Res 24:1020–1027
D’Astolfo DS, Pagliero RJ, Pras A et al (2015) Efficient intracellular delivery of native proteins. Cell 161:674–690
Zuris JA, Thompson DB, Shu Y et al (2015) Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. Nat Biotechnol 33:73–80
Woo JW, Kim J, Kwon SI et al (2015) DNA-free genome editing in plants with preassembled CRISPR-Cas9 ribonucleoproteins. Nat Biotechnol 33:1162–1164
Kimberland ML, Hou W, Alfonso-Pecchio A et al (2018) Strategies for controlling CRISPR/Cas9 off-target effects and biological variations in mammalian genome editing experiments. J Biotechnol 284:91–101
Lieber MR (2010) The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu Rev Biochem 79:181–211
Maruyama T, Dougan SK, Truttmann MC et al (2015) Increasing the efficiency of precise genome editing with CRISPR-Cas9 by inhibition of nonhomologous end joining. Nat Biotechnol 33:538–542
Ding Q, Regan SN, Xia Y et al (2013) Enhanced efficiency of human pluripotent stem cell genome editing through replacing TALENs with CRISPRs. Cell Stem Cell 12:393–394
Mandal PK, Ferreira LMR, Collins R et al (2014) Efficient ablation of genes in human hematopoietic stem and effector cells using CRISPR/Cas9. Cell Stem Cell 15:643–652
Zhang L, Jia R, Palange NJ et al (2015) Large genomic fragment deletions and insertions in mouse using CRISPR/Cas9. PLoS One 10:e0120396
Zhu S, Li W, Liu J et al (2016) Genome-scale deletion screening of human long non-coding RNAs using a paired-guide RNA CRISPR-Cas9 library. Nat Biotechnol 34:1279–1286
Cheung AH-K, Chow C, Zhang J et al (2018) Specific targeting of point mutations in EGFR L858R-positive lung cancer by CRISPR/Cas9. Lab Investig 98:968–976
Kim W, Lee S, Kim HS et al (2018) Targeting mutant KRAS with CRISPR-Cas9 controls tumor growth. Genome Res. https://doi.org/10.1101/gr.223891.117
Jin K, Pandey NB, Popel AS (2017) Crosstalk between stromal components and tumor cells of TNBC via secreted factors enhances tumor growth and metastasis. Oncotarget 8:60210–60222. https://doi.org/10.18632/oncotarget.19417
Liao L, Song M, Li X et al (2017) E3 ubiquitin ligase UBR5 drives the growth and metastasis of triple-negative breast cancer. Cancer Res 77:2090–2101
Bu X, Kato J, Hong JA et al (2018) CD38 knockout suppresses tumorigenesis in mice and clonogenic growth of human lung cancer cells. Carcinogenesis 39:242–251
Wang H, Yang H, Shivalila CS et al (2013) One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 153:910–918
Hendriks WT, Warren CR, Cowan CA (2016) Genome editing in human pluripotent stem cells: approaches, pitfalls, and solutions. Cell Stem Cell 18:53–65
Radecke S, Radecke F, Cathomen T, Schwarz K (2010) Zinc-finger nuclease-induced gene repair with oligodeoxynucleotides: wanted and unwanted target locus modifications. Mol Ther 18:743–753
Hendel A, Kildebeck EJ, Fine EJ et al (2014) Quantifying genome-editing outcomes at endogenous loci with SMRT sequencing. Cell Rep 7:293–305
Richardson CD, Ray GJ, DeWitt MA et al (2016) Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. Nat Biotechnol 34:339–344
Liang X, Potter J, Kumar S et al (2017) Enhanced CRISPR/Cas9-mediated precise genome editing by improved design and delivery of gRNA, Cas9 nuclease, and donor DNA. J Biotechnol 241:136–146
Chu VT, Weber T, Wefers B et al (2015) Increasing the efficiency of homology-directed repair for CRISPR-Cas9-induced precise gene editing in mammalian cells. Nat Biotechnol 33:543–548
Song J, Yang D, Xu J et al (2016) RS-1 enhances CRISPR/Cas9- and TALEN-mediated knock-in efficiency. Nat Commun 7:10548
Lin S, Staahl BT, Alla RK, Doudna JA (2014) Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. Elife 3:e04766
Gutschner T, Haemmerle M, Genovese G et al (2016) Post-translational regulation of Cas9 during G1 enhances homology-directed repair. Cell Rep 14:1555–1566
Aird EJ, Lovendahl KN, St Martin A et al (2018) Increasing Cas9-mediated homology-directed repair efficiency through covalent tethering of DNA repair template. Commun Biol 1:54
Savic N, Ringnalda FC, Lindsay H et al (2018) Covalent linkage of the DNA repair template to the CRISPR-Cas9 nuclease enhances homology-directed repair. Elife 7. https://doi.org/10.7554/eLife.33761
Nishida K, Arazoe T, Yachie N et al (2016) Targeted nucleotide editing using hybrid prokaryotic and vertebrate adaptive immune systems. Science:353. https://doi.org/10.1126/science.aaf8729
Suzuki K, Tsunekawa Y, Hernandez-Benitez R et al (2016) In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature 540:144–149
Suzuki K, Izpisua Belmonte JC (2018) In vivo genome editing via the HITI method as a tool for gene therapy. J Hum Genet 63:157–164
Xie C, Chen Y-L, Wang D-F et al (2017) SgRNA expression of CRIPSR-Cas9 system based on MiRNA polycistrons as a versatile tool to manipulate multiple and tissue-specific genome editing. Sci Rep 7:5795
Powell SK, Rivera-Soto R, Gray SJ (2015) Viral expression cassette elements to enhance transgene target specificity and expression in gene therapy. Discov Med 19:49–57
Lee AY, Cho M-H, Kim S (2019) Recent advances in aerosol gene delivery systems using non-viral vectors for lung cancer therapy. Expert Opin Drug Deliv 16:757–772
Papadakis ED, Nicklin SA, Baker AH, White SJ (2004) Promoters and control elements: designing expression cassettes for gene therapy. Curr Gene Ther 4:89–113
Ma H, Wu Y, Dang Y et al (2014) Pol III promoters to express small RNAs: delineation of transcription initiation. Mol Ther Nucleic Acids 3:e161
Ma H, Sumbilla CM, Farrance IKG et al (2004) Cell-specific expression of SERCA, the exogenous Ca2+ transport ATPase, in cardiac myocytes. Am J Physiol, Cell Physiol 286:C556–C564
Evergren E, Benfenati F, Shupliakov O (2007) The synapsin cycle: a view from the synaptic endocytic zone. J Neurosci Res 85:2648–2656
Prasad K-MR XY, Yang Z et al (2011) Robust cardiomyocyte-specific gene expression following systemic injection of AAV: in vivo gene delivery follows a Poisson distribution. Gene Ther 18:43–52
Lee Y, Messing A, Su M, Brenner M (2008) GFAP promoter elements required for region-specific and astrocyte-specific expression. Glia 56:481–493
Davidoff MS, Middendorff R, Köfüncü E et al (2002) Leydig cells of the human testis possess astrocyte and oligodendrocyte marker molecules. Acta Histochem 104:39–49
Danielyan L, Tolstonog G, Traub P et al (2007) Colocalization of glial fibrillary acidic protein, metallothionein, and MHC II in human, rat, NOD/SCID, and nude mouse skin keratinocytes and fibroblasts. J Invest Dermatol 127:555–563
Hioki H, Kameda H, Nakamura H et al (2007) Efficient gene transduction of neurons by lentivirus with enhanced neuron-specific promoters. Gene Ther 14:872–882
Cahoy JD, Emery B, Kaushal A et al (2008) A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci 28:264–278
Prasad K-MR, Yang Z, Xu Y et al (2006) 30. Cardiac-specific gene expression using pseudotyped AAV vectors and the cardiac troponin-T promoter. Molecular therapy 13:S13. https://doi.org/10.1016/j.ymthe.2006.08.042
Lee C-J, Fan X, Guo X, Medin JA (2011) Promoter-specific lentivectors for long-term, cardiac-directed therapy of Fabry disease. J Cardiol 57:115–122
Gou D, Narasaraju T, Chintagari NR et al (2004) Gene silencing in alveolar type II cells using cell-specific promoter in vitro and in vivo. Nucleic Acids Res 32:e134
Yamashita MS de A, Melo EO (2018) Mucin 2 (MUC2) promoter characterization: an overview. Cell Tissue Res 374:455–463
Igarashi P, Shashikant CS, Thomson RB et al (1999) Ksp-cadherin gene promoter. II. Kidney-specific activity in transgenic mice. Am J Phys 277:F599–F610
Wooddell CI, Reppen T, Wolff JA, Herweijer H (2008) Sustained liver-specific transgene expression from the albumin promoter in mice following hydrodynamic plasmid DNA delivery. J Gene Med 10:551–563
Iwata M, Englund DA, Wen Y et al (2018) A novel tetracycline-responsive transgenic mouse strain for skeletal muscle-specific gene expression. Skelet Muscle 8:33
Hamilton-Williams EE, Palmer SE, Charlton B, Slattery RM (2003) Beta cell MHC class I is a late requirement for diabetes. Proc Natl Acad Sci U S A 100:6688–6693
Wang Y, Rajala A, Cao B et al (2016) Cell-specific promoters enable lipid-based nanoparticles to deliver genes to specific cells of the retina in vivo. Theranostics 6:1514–1527
Dow LE, Fisher J, O’Rourke KP et al (2015) Inducible in vivo genome editing with CRISPR-Cas9. Nat Biotechnol 33:390–394
Cao J, Wu L, Zhang S-M et al (2016) An easy and efficient inducible CRISPR/Cas9 platform with improved specificity for multiple gene targeting. Nucleic Acids Res 44:e149
de Solis CA, Ho A, Holehonnur R, Ploski JE (2016) The development of a viral mediated CRISPR/Cas9 system with doxycycline dependent gRNA expression for inducible in vitro and in vivo genome editing. Front Mol Neurosci 9:70
Kumar N, Stanford W, de Solis C et al (2018) The development of an AAV-based CRISPR SaCas9 genome editing system that can be delivered to neurons in vivo and regulated via doxycycline and Cre-recombinase. Front Mol Neurosci 11:413
Tang W, Hu JH, Liu DR (2017) Aptazyme-embedded guide RNAs enable ligand-responsive genome editing and transcriptional activation. Nat Commun 8:15939
Oakes BL, Nadler DC, Flamholz A et al (2016) Profiling of engineering hotspots identifies an allosteric CRISPR-Cas9 switch. Nat Biotechnol 34:646–651
Davis KM, Pattanayak V, Thompson DB et al (2015) Small molecule-triggered Cas9 protein with improved genome-editing specificity. Nat Chem Biol 11:316–318
Maji B, Moore CL, Zetsche B et al (2017) Multidimensional chemical control of CRISPR-Cas9. Nat Chem Biol 13:9–11
Senturk S, Shirole NH, Nowak DG et al (2017) Rapid and tunable method to temporally control gene editing based on conditional Cas9 stabilization. Nat Commun 8:14370
Richter F, Fonfara I, Bouazza B et al (2016) Engineering of temperature- and light-switchable Cas9 variants. Nucleic Acids Res 44:10003–10014
Mullokandov G, Baccarini A, Ruzo A et al (2012) High-throughput assessment of microRNA activity and function using microRNA sensor and decoy libraries. Nat Methods 9:840–846
Shin J, Jiang F, Liu J-J et al (2017) Disabling Cas9 by an anti-CRISPR DNA mimic. Sci Adv 3:e1701620
Dong D, Guo M, Wang S et al (2017) Structural basis of CRISPR-SpyCas9 inhibition by an anti-CRISPR protein. Nature 546:436–439
Rauch BJ, Silvis MR, Hultquist JF et al (2017) Inhibition of CRISPR-Cas9 with bacteriophage proteins. Cell 168:150–158.e10
Yang H, Patel DJ (2017) Inhibition mechanism of an anti-CRISPR suppressor AcrIIA4 targeting SpyCas9. Mol cell 67:117–127.e5
He L, Hannon GJ (2004) MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet 5:522–531
Hirosawa M, Fujita Y, Parr CJC et al (2017) Cell-type-specific genome editing with a microRNA-responsive CRISPR-Cas9 switch. Nucleic Acids Res 45:e118
Hirosawa M, Fujita Y, Saito H (2019) Cell-type-specific CRISPR activation with microRNA-responsive AcrllA4 switch. ACS Synth Biol 8:1575–1582
Hoffmann MD, Aschenbrenner S, Grosse S et al (2019) Cell-specific CRISPR-Cas9 activation by microRNA-dependent expression of anti-CRISPR proteins. Nucleic Acids Res 47:e75
Wang X-W, Hu L-F, Hao J et al (2019) A microRNA-inducible CRISPR-Cas9 platform serves as a microRNA sensor and cell-type-specific genome regulation tool. Nat Cell Biol 21:522–530
Greene SC, Halmer T, Carey JM et al (2018) Theophylline toxicity: an old poisoning for a new generation of physicians. Turk J Emerg Med 18:37–39
Hacein-Bey-Abina S, Garrigue A, Wang GP et al (2008) Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J Clin Invest 118:3132–3142
Nayak S, Herzog RW (2010) Progress and prospects: immune responses to viral vectors. Gene Ther 17:295–304
Kay MA (2011) State-of-the-art gene-based therapies: the road ahead. Nat Rev Genet 12:316–328
Dong B, Nakai H, Xiao W (2010) Characterization of genome integrity for oversized recombinant AAV vector. Mol Ther 18:87–92
Senís E, Fatouros C, Große S et al (2014) CRISPR/Cas9-mediated genome engineering: an adeno-associated viral (AAV) vector toolbox. Biotechnol J 9:1402–1412
Swiech L, Heidenreich M, Banerjee A et al (2015) In vivo interrogation of gene function in the mammalian brain using CRISPR-Cas9. Nat Biotechnol 33:102–106
Nishimasu H, Ran FA, Hsu PD et al (2014) Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell 156:935–949
Chew WL, Tabebordbar M, Cheng JKW et al (2016) A multifunctional AAV-CRISPR-Cas9 and its host response. Nat Methods 13:868–874
Pack DW, Hoffman AS, Pun S, Stayton PS (2005) Design and development of polymers for gene delivery. Nat Rev Drug Discov 4:581–593
Lee K, Conboy M, Park HM et al (2017) Nanoparticle delivery of Cas9 ribonucleoprotein and donor DNA in vivo induces homology-directed DNA repair. Nat Biomed Eng 1:889–901
Wang M, Zuris JA, Meng F et al (2016) Efficient delivery of genome-editing proteins using bioreducible lipid nanoparticles. Proc Natl Acad Sci U S A 113:2868–2873
Li L, He Z-Y, Wei X-W et al (2015) Challenges in CRISPR/CAS9 delivery: potential roles of nonviral vectors. Hum Gene Ther 26:452–462
Ramamoorth M, Narvekar A (2015) Non viral vectors in gene therapy- an overview. J Clin Diagn Res 9:GE01–GE06
Ginn SL, Amaya AK, Alexander IE et al (2018) Gene therapy clinical trials worldwide to 2017: an update. J Gene Med 20:e3015
Steichen SD, Caldorera-Moore M, Peppas NA (2013) A review of current nanoparticle and targeting moieties for the delivery of cancer therapeutics. Eur J Pharm Sci 48:416–427
Bazak R, Houri M, El Achy S et al (2015) Cancer active targeting by nanoparticles: a comprehensive review of literature. J Cancer Res Clin Oncol 141:769–784
Sudimack J, Lee RJ (2000) Targeted drug delivery via the folate receptor. Adv Drug Deliv Rev 41:147–162
He Z-Y, Zhang Y-G, Yang Y-H et al (2018) In vivo ovarian cancer gene therapy using CRISPR-Cas9. Hum Gene Ther 29:223–233
Deshpande P, Jhaveri A, Pattni B et al (2018) Transferrin and octaarginine modified dual-functional liposomes with improved cancer cell targeting and enhanced intracellular delivery for the treatment of ovarian cancer. Drug Deliv 25:517–532
Xin H, Jiang X, Gu J et al (2011) Angiopep-conjugated poly(ethylene glycol)-co-poly(ε-caprolactone) nanoparticles as dual-targeting drug delivery system for brain glioma. Biomaterials 32:4293–4305
Kim JS, Shin DH, Kim J-S (2018) Dual-targeting immunoliposomes using angiopep-2 and CD133 antibody for glioblastoma stem cells. J Control Release 269:245–257
Papademetriou I, Vedula E, Charest J, Porter T (2018) Effect of flow on targeting and penetration of angiopep-decorated nanoparticles in a microfluidic model blood-brain barrier. PLoS One 13:e0205158
Liang C, Li F, Wang L et al (2017) Tumor cell-targeted delivery of CRISPR/Cas9 by aptamer-functionalized lipopolymer for therapeutic genome editing of VEGFA in osteosarcoma. Biomaterials 147:68–85
Affiliated Hospital to Academy of Military Medical Sciences, Peking University, Capital Medical University Safety of Transplantation of CRISPR CCR5 Modified CD34+ Cells in HIV-infected Subjects with Hematological Malignances. In: NLM Identifier: NCT03164135. https://clinicaltrials.gov/show/NCT03164135. Accessed 8 Aug 2019
Xu L, Yang H, Gao Y et al (2017) CRISPR/Cas9-mediated CCR5 ablation in human hematopoietic stem/progenitor cells confers HIV-1 resistance in vivo. Mol Ther 25:1782–1789
Zischewski J, Fischer R, Bortesi L (2017) Detection of on-target and off-target mutations generated by CRISPR/Cas9 and other sequence-specific nucleases. Biotechnol Adv 35:95–104
Li J, Manghwar H, Sun L et al (2019) Whole genome sequencing reveals rare off-target mutations and considerable inherent genetic or/and somaclonal variations in CRISPR /Cas9-edited cotton plants. Plant Biotechnol J 17:858–868
Jiang Y, Jiang Y, Wang S et al (2019) Optimal sequencing depth design for whole genome re-sequencing in pigs. BMC Bioinformatics 20:556
Akcakaya P, Bobbin ML, Guo JA et al (2018) In vivo CRISPR editing with no detectable genome-wide off-target mutations. Nature 561:416–419
Tsai SQ, Nguyen NT, Malagon-Lopez J et al (2017) CIRCLE-seq: a highly sensitive in vitro screen for genome-wide CRISPR–Cas9 nuclease off-targets. Nat Methods 14:607–614
van der Stegen SJC, Hamieh M, Sadelain M (2015) The pharmacology of second-generation chimeric antigen receptors. Nat Rev Drug Discov 14:499–509
Jensen MC, Riddell SR (2015) Designing chimeric antigen receptors to effectively and safely target tumors. Curr Opin Immunol 33:9–15
Baylor College of Medicine, The Methodist Hospital System, Texas Children’s Hospital, Center for Cell and Gene Therapy, B. C. o. M. Cell therapy for high risk T-cell malignancies using CD7-specific CAR expressed on autologous T cells. In: NLM Identifier: NCT03690011. https://clinicaltrials.gov/show/NCT03690011. Accessed 8 Aug 2019
Gomes-Silva D, Srinivasan M, Sharma S et al (2017) CD7-edited T cells expressing a CD7-specific CAR for the therapy of T-cell malignancies. Blood 130:285–296
Long C, Amoasii L, Mireault AA et al (2016) Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science 351:400–403
Nelson CE, Hakim CH, Ousterout DG et al (2016) In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science 351:403–407
Tabebordbar M, Zhu K, Cheng JKW et al (2016) In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science 351:407–411
Bengtsson NE, Hall JK, Odom GL et al (2017) Muscle-specific CRISPR/Cas9 dystrophin gene editing ameliorates pathophysiology in a mouse model for Duchenne muscular dystrophy. Nat Commun 8:14454
Kim K, Park SW, Kim JH et al (2017) Genome surgery using Cas9 ribonucleoproteins for the treatment of age-related macular degeneration. Genome Res 27:419–426
Ruan G-X, Barry E, Yu D et al (2017) CRISPR/Cas9-mediated genome editing as a therapeutic approach for Leber congenital Amaurosis 10. Mol Ther 25:331–341
Gao X, Tao Y, Lamas V et al (2018) Treatment of autosomal dominant hearing loss by in vivo delivery of genome editing agents. Nature 553:217–221
Raper SE, Chirmule N, Lee FS et al (2003) Fatal systemic inflammatory response syndrome in a ornithine transcarbamylase deficient patient following adenoviral gene transfer. Mol Genet Metab 80:148–158
Lander ES, Baylis F, Zhang F et al (2019) Adopt a moratorium on heritable genome editing. Nature 567:165–168
Funding
This work is supported by the Agency for Science, Technology and Research (Singapore) and by a grant from the National Medical Research Council to Boon-Seng Soh (NMRC/OFYIRG/0017/2016). Jeremy Kah Sheng Pang is supported by the National University of Singapore graduate scholarship.
Author information
Authors and Affiliations
Contributions
Hua Alexander Han, Jeremy Kah Sheng Pang and Boon-Seng Soh wrote the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Han, H.A., Pang, J.K.S. & Soh, BS. Mitigating off-target effects in CRISPR/Cas9-mediated in vivo gene editing. J Mol Med 98, 615–632 (2020). https://doi.org/10.1007/s00109-020-01893-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-020-01893-z