

Abstract
Background/Aim: Non-small cell lung cancer patients with epidermal growth factor receptor (EGFR) mutation have been shown to have a good response to erlotinib, a receptor tyrosine kinase inhibitor of EGFR. In this study, we found that the cell death pathways activated by erlotinib in 2D and 3D culture systems are different. Materials and Methods: The cell death pathways induced by erlotinib were evaluated by flow cytometry and immunoblotting in both 2D and 3D culture systems of EGFR mutant lung cancer cells. Results: Treatment with erlotinib induced caspase 8 activation and up-regulation of TNF-related apoptosis-inducing ligand (TRAIL) expression only in 3D cultures. Knockdown of TRAIL attenuated both erlotinib-induced activation of caspase-8 and apoptosis in 3D cultures. Erlotinib also increased LC3, an autophagy marker, expression and c-Jun N terminal kinase (JNK) activation. Both 3-MA as an autophagy inhibitor and SP600125 as a JNK inhibitor, significantly inhibited erlotinib-induced cell death. Conclusion: Erlotinib induces apoptotic cell death in 3D cultures through an autophagy-TRAIL-JNK pathway.
- Erlotinib
- 3D spheroid
- TRAIL
- autophagy
- lung cancer
Lung cancer was the most common cause of cancer-related death in males and the second most common cause of death in females world-wide in 2018 (1). Receptor tyrosine kinases such as epidermal growth factor receptor (EGFR) and anaplastic lymphoma kinase (ALK) are dysregulated and oncogenic in some non-small cell lung cancer (NSCLC) cases (2, 3). Among these tyrosine kinases, EGFR has well-characterized roles in cell proliferation, survival, invasion, and metastasis, through gene amplification or gain-of-function mutations, such as the T790 mutation in the EGFR kinase domain (4, 5). This dysregulation of EGFR results in oncogenic transformation, and patients with mutations in this gene have a poorer survival rate than patients without mutations (6).
Erlotinib, which inhibits tyrosine kinase activity by binding to the ATP-binding pocket of EGFR, has been approved and is now widely used to treat NSCLC patients with EGFR mutation (7). Molecular mechanisms underlying cell death by erlotinib have been described for standard 2D monolayer NSCLC cultures. In this setting, EGFR inhibition by erlotinib induces Bcl-2 interacting mediator of cell death (BIM)- or reactive-oxygen species (ROS)-dependent apoptotic cell death (8, 9). However, in 3D NSCLC cell cultures, the mechanism by which erlotinib induces cell death has yet to be explored, even though this information could potentially inform novel anti-cancer therapies for NSCLC patients.
Three-dimensional spheroid cell culture systems, which have been shown to mimic the in vivo setting and resemble the tumor microenvironment better than 2D culture systems, have been utilized as anti-cancer drug testing platforms and a screening system for several cancers (10, 11). These studies found that 2D lung and mesothelioma cancer cells were generally more sensitive to typical anti-cancer chemo-agents or targeted agents than the corresponding 3D cells (12-14). However, whether the mechanisms of cell death by anti-cancer drugs differ between 2D and 3D cell culture systems is not well understood.
Autophagy, a cell degradation pathway that has either a tumor suppressor or oncogenic role in various cancer cells (15, 16), has been reported to play an important role in EGFR inhibitor-induced cell death (17, 18). Chen et al. suggested that autophagy initiated by gefitinib, another EGFR inhibitor, up-regulated TRAIL and induced apoptotic cell death (17). However, another group found that autophagy suppressed erlotinib-induced cell death in NSCLC. Autophagy, which has been shown to be enhanced in the 3D setting (17), could play a role in EGFR inhibition and cell death in 3D, but not 2D systems.
In this study, we investigated the mechanisms of cell death by erlotinib in NSCLC cells with EGFR mutations grown under 2D and 3D culture conditions. We found that erlotinib activates an autophagy-TRAIL-JNK pathway in the 3D culture system, but not in the 2D culture system. In other words, the cell death pathway activated by erlotinib differs between 2D and 3D culture systems.
Materials and Methods
Cell culture and reagents. NSCLC cell lines, HCC827 and HCC4006, were purchased from ATCC (American Type Culture Collection, Manassas, VA, USA). Cells were maintained in RPMI-1640 medium (Mediatech Inc., Corning Subsidiary, Manassas, VA, USA) containing heat-inactivated 10% fetal bovine serum (FBS; Tissue Culture Biologicals, Tulare, CA, USA) and 100 μg/ml penicillin/streptomycin (Sigma, St. Louis, MO, USA) in 5% CO2 at 37°C. Erlotinib was purchased from Selleck (Selleck Chemicals, Houston, TX, USA). Z-VAD-fmk (z-VAD), a pan-caspase inhibitor; z-IETD-fmk (z-IETD), a caspase-8 specific inhibitor; and SP600125, a JNK inhibitor, were purchased from Sigma.
3D spheroid cell culture. Cells were cultured in poly-HEMA-coated wells as reported previously (19). Briefly, HCC827 and HCC4006 cells were plated at a density of 1-2×103 cells per well in poly-HEMA (2-hydroxyethyl methacrylate, Sigma)-coated 96-well plates. Cells were incubated for 72 h until the formation of spheroids.
Lentiviral shRNA infection. To knock-down TRAIL expression, we used the pLKO1-based lentiviral system (Sigma). Briefly, HEK293T cells were transfected with lentiviral packaging mix (Sigma) and pLKO-1 scrambled or TRAIL shRNA (sc shRNA: 5’-AAT TCT CCG AAC GTG TCA CGT-3’; TRAIL shRNA: 5’-AAC GAG CUG AAG CAG AUG CAG-3’) using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA), according to the manufacturer’s instructions. HCC827 and HCC4006 cells were grown as 2D monolayers for 24 h, infected with 20 MOI (multiplicity of infection) pLKO-1 scrambled or TRAIL shRNA lentivirus for 24 h, and then re-seeded in poly-HEMA-coated 60 mm plates. After spheroid formation (72 h), cells were treated with 500 nM erlotinib for another 24 h. Validation of shRNAs was performed by Genolution (Genolution Pharmaceutical Inc., Seoul, Republic of Korea).
Cell death and Annexin-V staining. For the 2D culture studies, cells were seeded at a density of 3×105 in 60 mm dishes for 24 h and treated with erlotinib at the indicated doses for 24 h. For 3D culture studies, cells were cultivated in poly-HEMA-coated 96-wells for 72 h until they formed spheroids and then, incubated with erlotinib for another 24 h. Cells were washed with PBS, trypsinized, and stained with trypan blue or annexin-V/PI double staining solution (BD Biosciences, San Jose, CA, USA), followed by fluorescence-activated cell sorting (BD Biosciences).
Immunoblot analysis. Cells were lysed with RIPA lysis buffer (Tech & Innovation, Kangwon, Republic of Korea) containing a protease and phosphatase inhibitor cocktail (Sigma). After cell lysis, supernatants were collected after centrifugation at 13,000 rpm for 20 min. Protein concentrations were determined using the Bradford assay (Bio-Rad, USA). Equal total amounts of cellular proteins (20 μg per lane) were separated using 8-15% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and were transferred to an Immobilon-PVDF membrane (Millipore Corporation, Billerica, MA, USA). Membranes were blocked with 5% skim-milk in TBS-T (Tech & Innovation) and probed with antibodies against cleaved caspase-8, cleaved caspase-9, cleaved caspase-3, TRAIL, Fas, phospho-JNK, JNK, LC3B (Cell Signaling Technology, Beverly, MA, USA), or β-actin (Santa Cruz Biotechnology, Santa Cruz, CA, USA). Primary antibodies were detected using horseradish peroxidase-conjugated anti-mouse or anti-rabbit secondary antibodies (Cell Signaling) and an enhanced chemiluminescence detection system (Amersham, Buckinghamshire, UK).
Statistical analysis. All data were shown as the mean±SD. Two ANOVA analysis using SPSS 26 (SPSS Inc., Chicago, IL, USA) were performed to evaluate p-values, and p-values less than 0.05 were considered significant.
Results
Erlotinib induces caspase-8-dependent apoptotic cell death in 3D culture but not in 2D culture. First, we investigated the effect of erlotinib on cell viability in 2D and 3D cultures, using HCC827 and HCC4006 cells, which have been reported to form 3D spheroids (20). It was reported that HCC827 and HCC4006 cells, which have EGFR gene amplification and deletion of exon 19, are sensitive to erlotinib (21). As has been reported previously (20), we found that phosphorylation of EGFR was dramatically up-regulated in 3D cultures compared to 2D cultures in both HCC827 and HCC4006 cells (Figure 1A and B). Moreover, as reported previously, the rate of cell death in NSCLC 2D culture cells increased significantly after exposure to erlotinib in a dose-dependent manner. Surprisingly, an increased rate of cell death was found in response to erlotinib in cells grown in 3D culture compared to 2D (Figure 1C and D).

Erlotinib induces cell death in both 2D and 3D NSCLC cell culture systems. Erlotinib inhibits the phosphorylation of EGFR in both 2D and 3D cultures. HCC827 and HCC4006 cells grown in 2D and 3D cultures were treated with erlotinib at the indicated doses. Cell lysates were prepared for immunoblot analysis using anti-p-EGFR or anti-EGFR antibodies. β-actin was used as a loading control (A and B). HCC827 and HCC4006 cells were treated with erlotinib at the indicated doses for 24 h. Cell death was determined by the trypan blue exclusion method. Error bars represent the mean±S.D. of three separate experiments performed in triplicate (C and D). 3D spheroid cells were pretreated with 50 μM z-VAD before exposure to 500 nM erlotinib; after 24 h, cell death was evaluated by the trypan blue method (E and F). Error bars represent mean±S.D. of three separate experiments. * and ** indicate p<0.05 and p<0.01, respectively.
Next, we evaluated whether cell death by erlotinib is caspase-dependent in 3D cultures, as has been described for NSCLC cells grown in 2D culture (21). We found that cell death by erlotinib was completely abrogated by pretreatment of cells grown in 2D and 3D culture with a pan-caspase inhibitor (Figure 1E and F). These results indicate that erlotinib treatment induces caspase-dependent apoptotic cell death in both 2D and 3D cultures.
Next, we examined the effect of erlotinib on the caspase cascade in 2D and 3D cultures. Interestingly, caspase-8 was activated after exposure to erlotinib in 3D- but not in 2D-cultured cells (Figure 2A and B). To confirm the activation of caspase-8 in 3D cell cultures, we tested whether treatment with a caspase-8 inhibitor, z-IETD, decreased erlotinib-induced cell death in 3D cultures but not in 2D cultures, and found that that this was the case (Figure 2C and D). These results suggest that erlotinib induces caspase-cascade apoptotic cell death differently in 2D and 3D cell cultures.

Erlotinib activates caspase-8 in 3D, not in the 2D, cell culture systems. (A and B) HCC827 and HCC4006 cells grown under 2D and 3D culture conditions were treated with 500 nM erlotinib for 24 h. Cell lysates were prepared for immunoblot analysis. β-actin was used as a loading control. (C and D) Cells were treated with 500 nM erlotinib after pre-treatment with 20 μM z-IETD, and cell death was measured by the trypan blue method. Error bars indicate mean±S.D. of three separate experiments. **p<0.01.
Erlotinib up-regulates the expression of TRAIL in 3D cell cultures only. As shown in Figure 2, cell death by erlotinib was associated with caspase-8 activation in 3D-cultured cells. Caspase-8 is an initiator caspase that is activated by receptor-mediated apoptosis, such as that induced by Fas, TRAIL, or TNF-α. Therefore, we examined the expression of TRAIL and Fas after erlotinib treatment. Interestingly, treatment with erlotinib induced the expression of TRAIL in 3D cell cultures but not in 2D cell cultures (Figure 3A and B). Next, we evaluated the effect of TRAIL expression on erlotinib-induced cell death in 3D cultures. We infected cells with lentiviral TRAIL shRNA and then, treated the cells with erlotinib. Down-regulation of TRAIL completely inhibited erlotinib-induced apoptosis, as measured by annexin-V positivity, in 3D cultures (Figure 3C and D). Consistently, after TRAIL shRNA treatment, cleavage of caspase-8 and caspase-3 was inhibited in 3D cultures exposed to erlotinib (Figure 3E and F). Together, these results demonstrate that erlotinib induces apoptosis in cells grown in 3D cultures in a TRAIL-dependent manner.
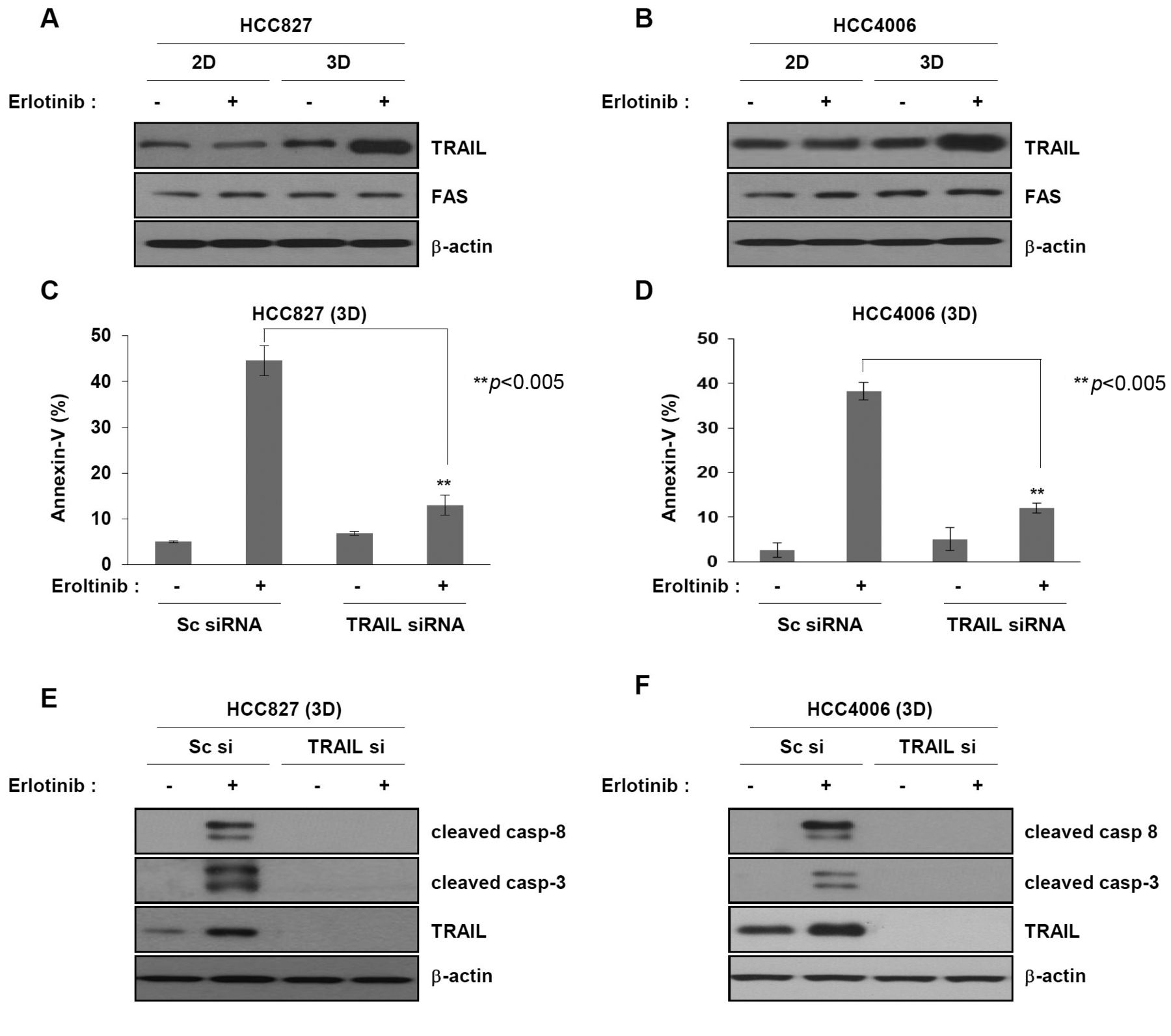
Erlotinib induces expression of TRAIL in 3D culture only. (A and B) HCC827 and HCC4006 cells grown under 2D and 3D culture conditions were treated with erlotinib for 24 h. Cell lysates were subjected to immunoblot analysis with TRAIL or anti-Fas antibodies. (C, D, E and F) HCC827 and HCC4006 cells grown under 3D culture conditions were infected with scrambled or TRAIL shRNA lentivirus for 24 h, re-seeded in poly-HEMA coated dish until spheroid formation at 72 h, and then treated with 500 nM erlotinib for 24 h. (C and D) Cell death was measured by annexin-V staining using FACS. Error bars represent mean±S.D. of three separate experiments. **p<0.01. (E and F) Expression of caspases and TRAIL was determined by immunoblot analysis using anti-cleaved caspase-8, anti-cleaved caspase-3, or anti-TRAIL. β-actin was used as a loading control.
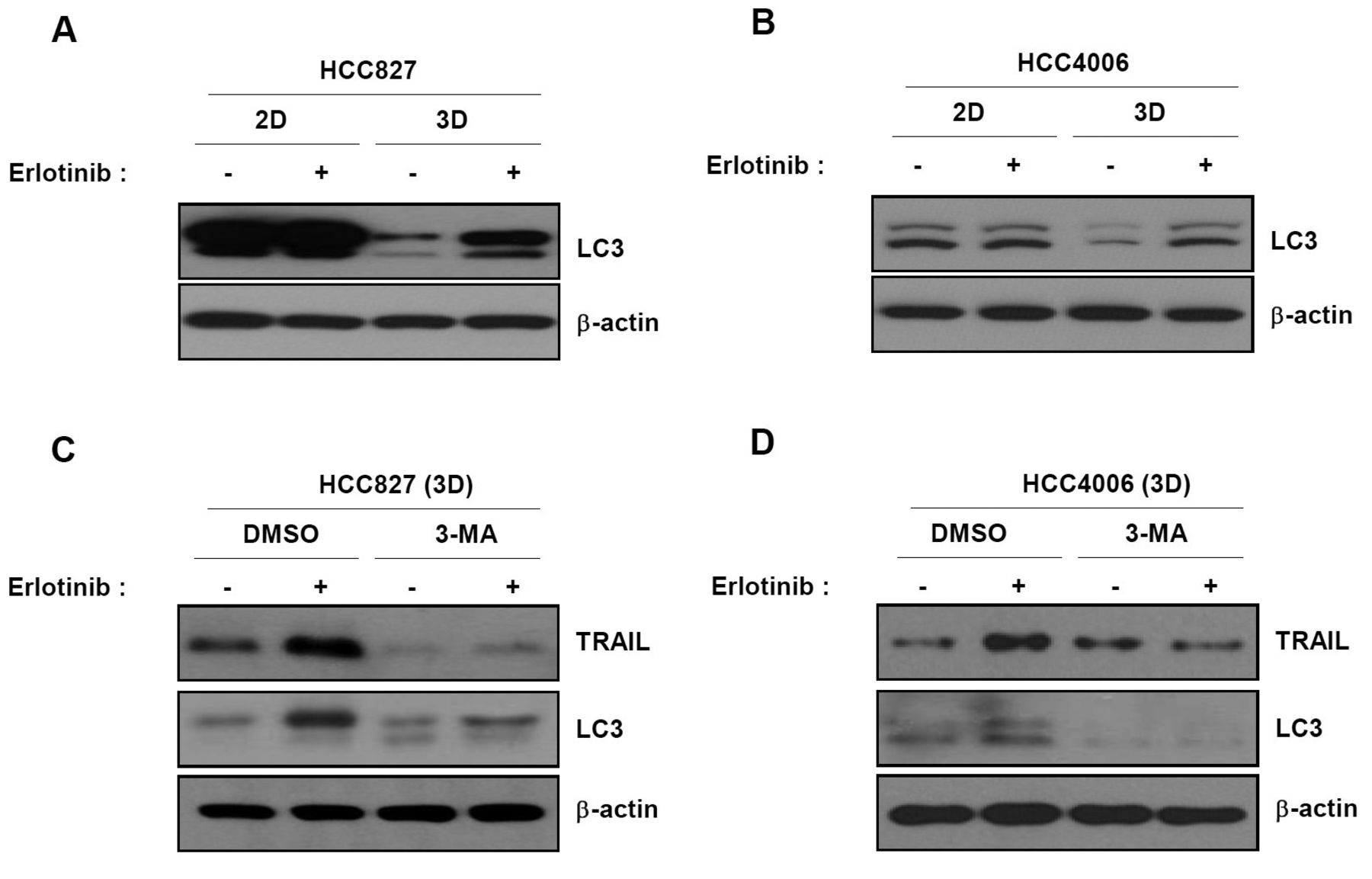
In 3D cultures, autophagy precedes activation of TRAIL after exposure to erlotinib. As shown in Figure 3, TRAIL is a key factor in cell death induction by erlotinib in 3D cultures. Accordingly, we investigated the molecular mechanisms underlying TRAIL up-regulation by erlotinib in 3D cultures. Autophagy was recently reported to be responsible for TRAIL activation following EGFR inhibition (18). Thus, we examined the effect of autophagy on the response to erlotinib. Levels of the LC3II protein, which is an autophagy marker, were dramatically increased in response to erlotinib treatment in 3D but not 2D cultures (Figure 4A and B). Next, we examined the effect of autophagy on the expression of TRAIL following erlotinib treatment. We treated cells with 3-MA, an autophagy chemical inhibitor, before treatment with erlotinib. Up-regulation of TRAIL by erlotinib in 3D cultures was completely inhibited by 3-MA (Figure 4C and D). These data indicate that erlotinib induces TRAIL-mediated apoptotic cell death through activation of autophagy in 3D cultures.
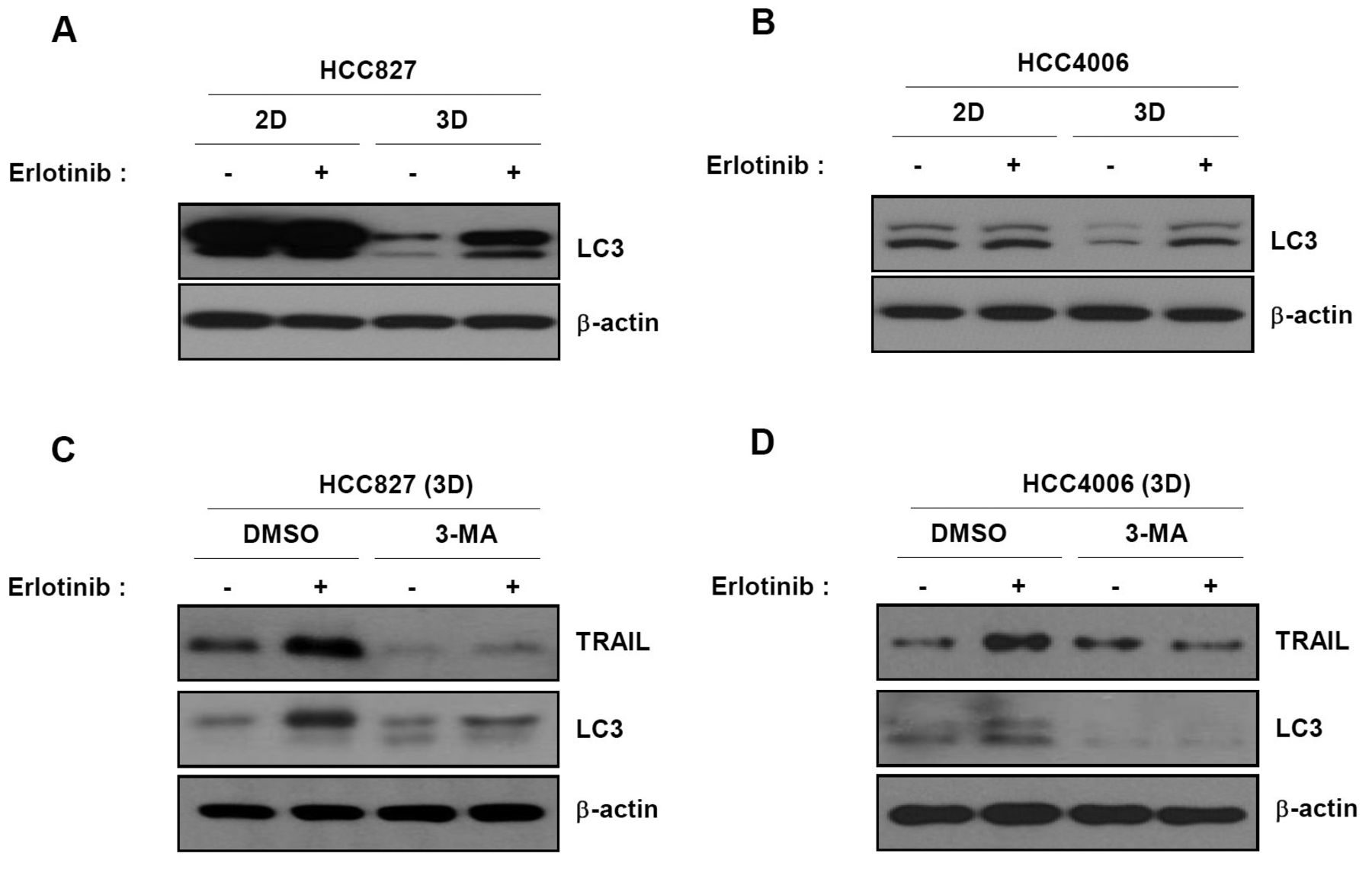
Up-regulation of TRAIL by erlotinib in 3D cultures is mediated by autophagy. (A and B) Cells were treated with erlotinib and subjected to immunoblot analysis using anti-LC3 as a marker of autophagy. (C and D) 3D spheroid cells were pre-treated with 3-MA, an autophagy inhibitor, for 1 h and then treated with erlotinib for another 24 h. Cell lysates were subjected to immunoblot analysis with anti-TRAIL or LC3 antibodies. Results are representative of three independent experiments.
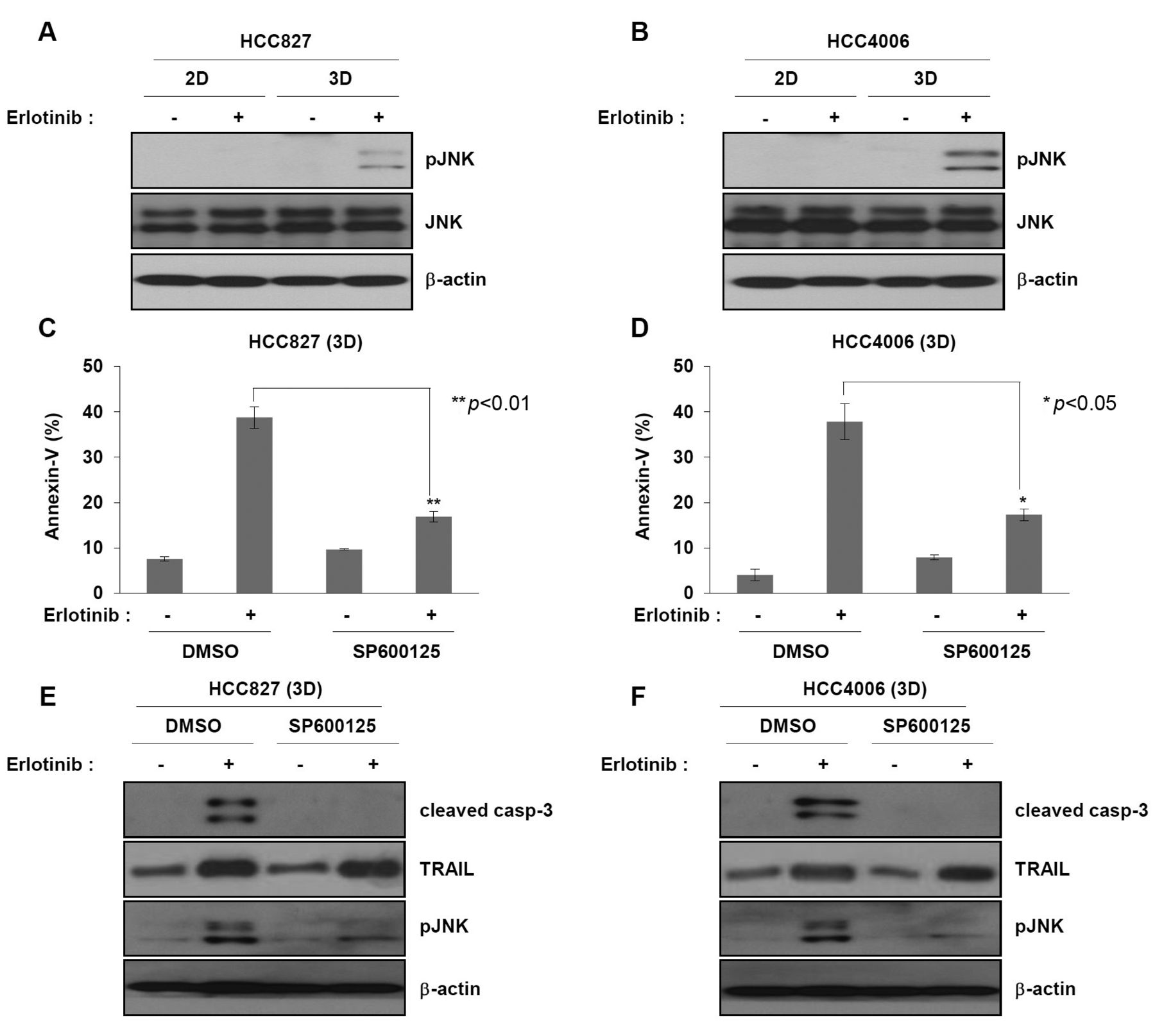
JNK plays an important role in erlotinib-induced cell death in 3D cultures. C-Jun N-terminal kinase (JNK) is associated with apoptosis induced by various death receptor ligands, including TRAIL and TNF-α (22, 23). To determine whether phosphorylation of JNK influenced cell death by erlotinib in 3D cultures, we evaluated the level of phosphorylated JNK in erlotinib-treated cultures. Phosphorylation of JNK occurred in erlotinib-treated 3D but not 2D cultures (Figure 5A and B). We further analyzed the effect of JNK phosphorylation on erlotinib-induced cell death using a JNK chemical inhibitor, SP600125. Treatment of 3D cultures with SP600125 blocked erlotinib-induced cell death (Figure 5C and D). Consistently, activation of caspase-3, but not activation of TRAIL, by erlotinib in 3D cultures was abrogated in SP600125-treated cells (Figure 5E and F). These results indicate that erlotinib-induced apoptotic cell death in 3D cultures likely involves sequential activation of TRAIL-JNK pathways.

JNK plays an important role in erlotinib-induced cell death of non-small cell lung cancer cells grown in 3D culture. (A and B) HCC827 and HCC4006 cells grown in 2D or 3D were treated with 500 nM erlotinib for 24 h. They were subjected to immunoblot analysis to determine the levels of phosphorylated JNK using anti-phospho JNK antibody. (C, D, E and F) HCC827 and HCC4006 3D spheroid cells were pretreated with 10 μM SP600125, a JNK chemical inhibitor, for 1 h before treatment with 500 nM erlotinib for 24 h. (C and D) Cell death was measured by annexin-V staining followed by FACS analysis. Graphs represent mean±S.D. of three separate experiments. **p<0.01. (E and F) Activation of caspase-3 and TRAIL was determined by immunoblot analysis using anti-cleaved caspase-3 or anti-TRAIL antibodies. Results are representative of three independent experiments.
Discussion
Prior studies have focused on the molecular mechanisms of erlotinib in NSCLC 2D cell culture systems (24, 25). However, the mechanism by which erlotinib induces cell death in NSCLC 3D culture systems has not been examined. Such studies might be clinically relevant because it is recognized that 3D cultures better reflect the situation in vivo (26, 27). Therefore, an understanding of the response to erlotinib in 3D cultures could result in the development of new clinical anti-cancer strategies for NSCLC patients. In this study, we found that the molecular mechanisms of erlotinib-induced cell death in 2D and 3D cultures differed.
It has been reported that tumor cells grown as 3D spheroids are more resistant to chemotherapeutic agents or targeted agents than 2D cells, and new combinational anti-cancer strategies for 3D spheroid tumor cells have been developed by studying the underlying molecular mechanisms (28-30). Based on these reports, we investigated the sensitivity of cells grown in 2D and 3D cultures to erlotinib. In contrast with previous studies, we found that NSCLC cells grown in 2D and 3D cultures showed no difference in sensitivity to erlotinib.
We then examined whether there was a difference in the cell death pathway activated by erlotinib in 2D and 3D cultures. Interestingly, the active form of caspase-8 was found only in 3D cultures after exposure to erlotinib. Moreover, blockade of caspase-8 resulted in resistance to erlotinib-induced cell death in 3D cultures only, suggesting that cell death in response to erlotinib is induced by different caspase cascades in 2D culture. In addition to TRAIL, TNF-α and Fas signaling pathways have been reported to initiate receptor-mediated apoptosis (31-33). We found that erlotinib elicited the expression of TRAIL, but not Fas, in 3D-cultured cells. Consistent with this observation, down-regulation of TRAIL completely abrogated erlotinib-induced caspase-8 activation and subsequent cell death in 3D cultures, implying that erlotinib induces apoptotic cell death via TRAIL-caspase-8 activation.
We further found that up-regulation of TRAIL by erlotinib was strongly associated with the induction of autophagy in 3D cultures. Treatment of colon cancer cells with gefitinib has been reported to induce autophagy and TRAIL expression (17). Another study reported that autophagy suppressed erlotinib-induced apoptotic cell death in NSCLC cells, implying that autophagy can inhibit apoptotic cell death (18). Interestingly, it was reported that autophagic flux in 3D, but not 2D, mesothelioma models better represented autophagy by the actual tumor; thus, the findings in 3D are likely to be more relevant to human disease (34). In our study, using 3D spheroids, erlotinib induced cell death by up-regulating TRAIL via autophagy. These findings suggest that up-regulation of autophagy or TRAIL might enhance the effect of erlotinib.
Several studies have reported that JNK acts as the main effector of TRAIL-induced apoptotic cell death in various cancers (23). On the basis of these reports, we investigated the role of JNK in erlotinib-induced cell death in 3D cultures. We observed that phosphorylation of JNK was dramatically increased in erlotinib-treated 3D cultures but not 2D cultures, and that erlotinib-induced apoptotic cell death was significantly decreased in JNK inhibitor-treated 3D culture cells, indicating that JNK plays a major role in erlotinib-induced apoptotic cell death in 3D cultures.
The 3D spheroid cell culture system has been proposed as an appropriate model of the in vivo tumor environment. In light of this, an understanding of the mechanism of action of erlotinib could facilitate development of novel anti-cancer therapies in NSCLC patients. Overall, our findings, as summarized in Figure 6, indicate that an autophagy-TRAIL-JNK pathway is involved in erlotinib-induced apoptosis of cells grown in 3D culture (Figure 6). Therefore, the effect of erlotinib in NSCLC patients can be enhanced by using TRAIL or other agonists, by activating JNK, or by stimulating autophagy. Further studies of erlotinib resistance in 3D cultured cells are needed to determine whether these approaches are helpful for forestalling or bypassing resistance to erlotinib.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Suggested mechanism of Erlotinib-induced apoptosis in non-small cell lung cancer (NSCLC) 2D/3D culture. Although erlotinib treatment induces caspase3-dependent apoptotic cell death in both 2D and 3D cultures according to previous other studies using NSCLC 2D cell culture systems, the activation caspase-8 was found only in 3D cultures. Overall, our findings indicate that an autophagy-TRAIL-JNK-caspase pathway is involved in erlotinib-induced apoptosis of cells grown in 3D culture.
Acknowledgements
This work was supported by the 2019 Inje University research grant. The Authors would like to thank Dario Barbone, PhD, for his assistance with preliminary studies.
Footnotes
This article is freely accessible online.
* These Authors contributed equally to this work.
Authors’ Contributions
Conceptualization, Hyun-Kyung Lee and Dae Young Hur; Data curation, Min Hye Noh, Seung-Woo Hong, and Dae Young Hur; Formal analysis, Seung-Woo Hong, Seung-Mi Kim and Sung Hyun Kim; Funding acquisition, Dae Young Hur; Investigation, Min Hye Noh, Seung-Woo Hong and Seung-Mi Kim; Project administration, Hyun-Kyung Lee, V. Courtney Broaddus and Dae Young Hur; Writing – original draft, Seung-Woo Hong and Yeong Seok Kim; Writing – review & editing, Hyun-Kyung Lee, V. Courtney Broaddus and Dae Young Hur.
Conflicts of Interest
The Authors declare no conflicts of interest in relation to this study.
- Received January 19, 2021.
- Revision received February 3, 2021.
- Accepted February 4, 2021.
- Copyright © 2021 International Institute of Anticancer Research (Dr. George J. Delinasios), All rights reserved.