

Abstract
Recently, therapeutic drug monitoring of 5-fluorouracil (5-FU), the key chemotherapeutic drug for colorectal cancer, has been applied in daily clinical practice and has contributed towards improving clinical outcomes. However, current dose modifications are based only on values of the area under the plasma concentration–time profile, which are simply calculated from plasma 5-FU concentrations and infusion periods. When dose-limiting toxicities occur, the dosing is empirically reduced or discontinued, leading to treatment failure. To prevent this predictable failure and obtain better clinical outcomes, rational dosage-based strategies are required for 5-FU. Combining therapeutic drug monitoring with a mathematical approach using a pharmacokinetic– pharmacodynamic/toxicodynamic model is expected to help simulate time-course profiles of the efficacy of drugs and the degree of toxicity, thereby contributing towards dose setting for individual patients. Therefore, to facilitate pharmacometric modelling and simulation techniques for optimising current oncology therapies, this review focuses on pharmacometrics approaches for personalizing 5-FU-based chemotherapy.
- Modelling and simulation
- chronopharmacokinetics
- translational research
- cancer chemotherapy
- therapeutic drug monitoring
- 5-FU prodrug
- pharmacometrics
- review
5-Fluorouracil (5-FU) has been used to treat patients for approximately 60 years and remains a cornerstone of colorectal cancer chemotherapy. The mechanism underlying the pharmacodynamic (PD) action of 5-FU involves its conversion to fluoro-deoxyuridine monophosphate (FdUMP), which subsequently inhibits thymidylate synthase via the formation of a ternary complex comprising FdUMP, thymidylate synthase, and 5,10-methylene tetrahydrofolate in tumour cells, consequently inhibiting DNA synthesis. 5-FU is also incorporated into RNA and prevents protein synthesis (1). After it reaches the blood circulation, over 80% of administered 5-FU is metabolized by dihydropyrimidine dehydrogenase (DPD), which is a rate-limiting enzyme, in the liver (2, 3).
An appropriate 5-FU administration schedule has been developed to improve clinical responses in the clinical oncology setting. A meta-analysis showed that the infusion schedule of 5-FU is superior to the bolus administration schedule with respect to response rate and overall survival (4). Currently, bolus plus long-term infusion schedules, such as the folinic acid/5-FU/irinotecan (FOLFIRI) and the folinic acid/5-FU/oxaliplatin (FOLFOX) regimens, are standard approaches for treating adjuvant or metastatic colorectal cancer. However, infusion regimens involve long hospital stays for patients and require catheterization. To overcome these shortcomings of infusion therapy, orally available prodrugs of 5-FU have been developed, such as capecitabine, tegafur/gimeracil/oteracil (S-1), and tegafur/uracil (UFT). The capecitabine plus oxaliplatin (XELOX), S-1 plus oxaliplatin, and UFT plus leucovorin regimens are accepted for clinical chemotherapy of colorectal cancer.
Various administration schedules have been developed for 5-FU-based chemotherapy; however, there are large inter- and intra-individual pharmacokinetic (PK) variabilities which are important contributors to clinical treatment failure, and the method for determining the optimal 5-FU dose is debatable. The standard approach for 5-FU dose determination is based on the body surface area (BSA); however, this approach leads to large, approximately 100-fold variability, inter-individual variability in plasma 5-FU level (5-7). Many clinical studies have shown that PK-guided dose adjustments of 5-FU can improve clinical efficacies and reduce toxicity (8-10). Therapeutic drug monitoring (TDM) of the plasma 5-FU level is recommended for personalization of 5-FU dosing to obtain adequate systemic exposure, leading to improved clinical efficacy and lesser adverse effects. In 2018, the academic members of the International Association of Therapeutic Drug Monitoring and Clinical Toxicology reviewed the use of TDM for 5-FU infusion and strongly recommended TDM of 5-FU in clinical practice (11). While TDM of 5-FU is an important tool for determining the optimal dose for individual patients and obtaining appropriate systemic exposure, additional challenges still remain for achieving personalized 5-FU-based chemotherapy and further improved clinical outcomes. During dose adjustments by TDM, for instance, the 5-FU dose in the first cycle must be determined on the basis of BSA, whereas the doses in all subsequent cycles are adjusted on the basis of the plasma 5-FU level achieved. Therefore, the optimum target concentration can only be achieved after some chemotherapy cycles are completed. Moreover, several patient characteristics (e.g. sex, age, body weight, tumour type, cancer stage, DPD phenotype and activity level, and co-administered drugs) and circadian fluctuations of plasma 5-FU concentration make it difficult to adequately estimate drug exposure and to determine individual dose setting during TDM. When dose-limiting toxicities are observed, empirical dose reduction or treatment discontinuation is required, which leads to treatment failure. Therefore, an approach that would provide the best quantitative prediction of drug exposure, therapeutic response, and toxicities on the basis of the plasma 5-FU concentration needs to be developed.
In recent years, the pharmacometrics approach has attracted widespread interest in the field of oncology for achieving personalized medicine in clinical practice (12). Pharmacometrics is defined as “the science of developing and applying mathematical and statistical methods to (a) characterize, understand, and predict a drug’s pharmacokinetic and pharmacodynamic behaviour; (b) quantify uncertainty of information about that behaviour; and (c) rationalize data-driven decision making in the drug development process and pharmacotherapy” (13). Pharmacometric modelling and simulation techniques can help understand the relationship between drug exposure and the subsequent effects, which is particularly important for determining the appropriate dose setting for each patient (14). Almost all anticancer agents have narrow therapeutic windows, and there are large inter- and intra-individual variabilities in drug exposure. A balance between drug efficacy and adverse events is required for obtaining the desired clinical outcomes, especially in the field of oncology. A mathematical method such as the use of PK– PD/toxicodynamic (PK-PD/TD) models as part of the pharmacometrics approach would be valuable for predicting the time course of profiles of plasma drug level and PD/TD responses such that the optimal dose schedule can be prescribed for the patient in order to maximize drug efficacy and minimize toxicity. Close collaboration between oncological physicians and pharmacometricians could lead to better prognosis on application of pharmacometrics in a clinical dose setting.
In this review, recent pharmacometrics approaches for the personalization of 5-FU-based chemotherapy in patients with cancer have been summarized. We have discussed pharmacometrics-related studies describing and simulating both 5-FU exposure and effects in various dose settings. This review focuses only on the PK-PD/TD model approach related to drug responses, including antitumor effects and dose-limiting toxicities, although there are many studies on classic compartmental PK models. Future perspectives on applying pharmacometrics to routinely collected clinical data and personalized medicine in order to optimize drug targeting are also discussed.
PK Model for Evaluating Circadian Variation
A circadian rhythm is observed in plasma 5-FU concentration during long-term infusion in clinical studies (15). Circadian fluctuations of plasma 5-FU level during constant infusion may be a major factor leading to incorrect PK and PD/TD estimation by the pharmacometrics approach; therefore, the current review also focused on the pharmacometrics approach in relation to the circadian rhythm of 5-FU PK. The circadian alterations in 5-FU PK might be derived from circadian variations in DPD activity and contribute to large inter- and intra-individual variations in plasma 5-FU concentration (16, 17). Conflicting results have been reported for the time points for peak and trough levels during the day (15). Harris et al. reported that plasma 5-FU levels obtained over a 24-h period reached peak values at 11:00 h and trough values at 23:00 h in patients who had cancer and were receiving continuous 5-FU infusion (300 mg m–2 d–1) (18). However, Metzger et al. reported that peak values were obtained at 04:00 h and trough values at 13:00 h in patients after 5-FU infusion (600 mg m–2 d–1) (19). Table I shows a previously reported PK model for evaluating and describing circadian changes in plasma 5-FU concentration. To the best of our knowledge, only one study has described a clinical PK model taking into account the circadian rhythm in plasma 5-FU concentration. Bressolle et al. defined the circadian model by the sum of two cosine cyclic components of 12- and 24-h periods and described circadian variations in 5-FU clearance (20). This circadian model was developed using 562 5-FU concentration datasets obtained for 65 patients and validated using another 104 datasets obtained for 20 patients. Analysis of this model revealed that the peak period for the plasma 5-FU concentration was approximately 04:00 h, consistent with previous results obtained by Metzger et al. (19). Moreover, the model was able to estimate 5-FU PK parameters for individual patients, thereby contributing to optimization of the dose regimen of each patient. On the basis of these findings, a chronomodulated chemotherapy regimen has been proposed (21-23). However, clinical application of chronomodulation of 5-FU dosing is limited in the current standard regimen, which may be attributable to difficulties in estimating the circadian rhythm of each patient.
Summary of pharmacokinetic (PK) model to describe circadian variations in plasma concentration of 5-fluorouracil (5-FU).
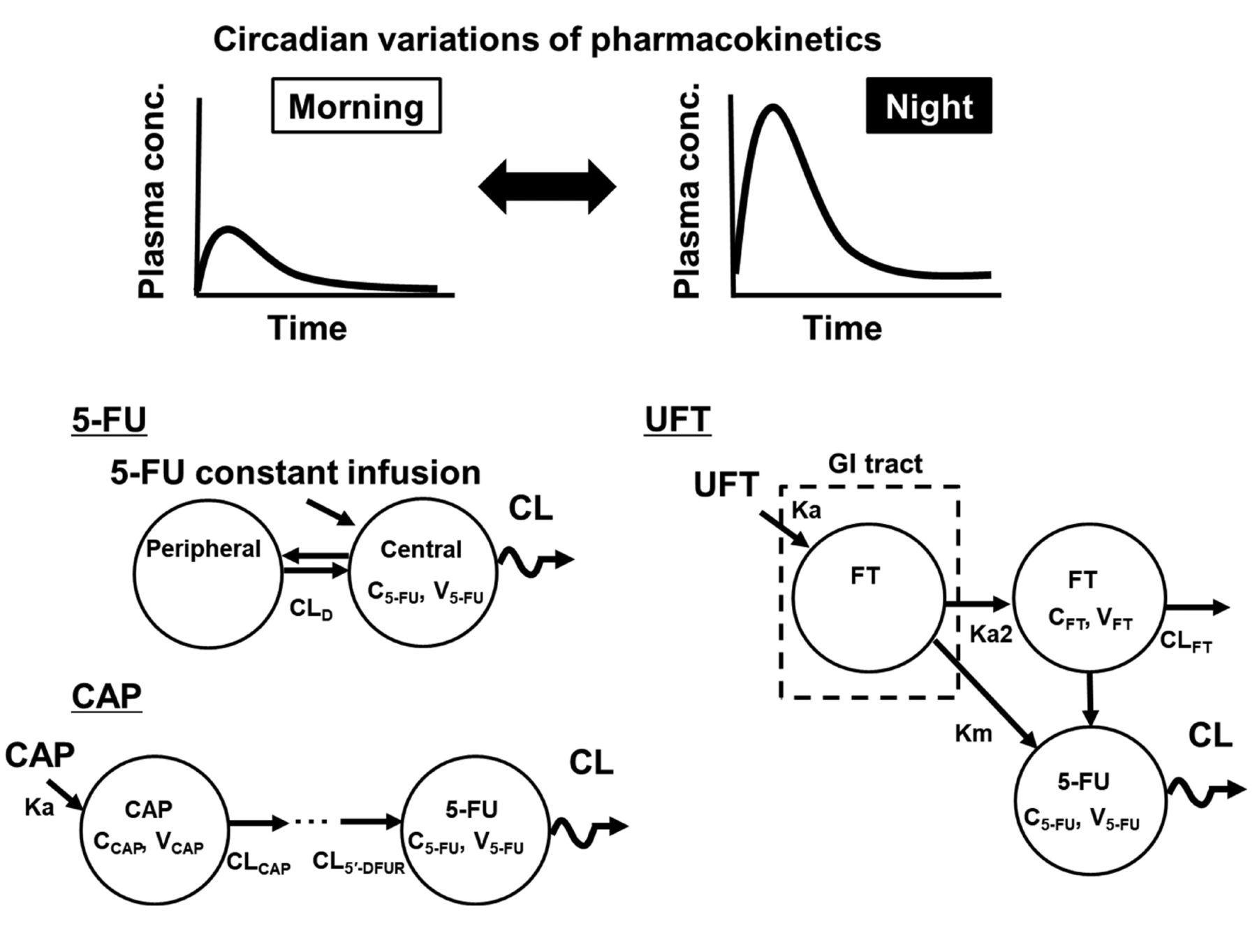
Various baseline characteristics in patients, including DPD activity level, may also affect circadian changes in plasma 5-FU levels. Recently, the circadian 5-FU patterns in animals were investigated using a PK model with the cosinor method (24-27) to exclude these potential contributing factors (Figure 1). In rats treated with continuous 5FU infusion (50 mg m–2 h–1) for 48 h, PK model analysis showed that the plasma 5-FU concentration followed a 24-h cosine circadian curve, representing an overall 1.8-fold increase from a nadir to a peak, with a relative amplitude (percentage of mesor) of 28% (27). Additionally, the loading bolus dose before initiating the infusion was found to contribute to circadian variations in plasma 5-FU level (27). These observations from animal studies suggest that in the recently modified regimen that omits bolus 5-FU injection, chronomodulation of dosing may enable sufficient clinical response with minimum toxicities and that timing of blood sampling during TDM procedures should be determined cautiously. These animal study findings indicate that further clinical evaluations using a PK model that takes into account circadian variations in plasma 5-FU level are required for deciding appropriate dosing schedules and blood sampling times in TDM.
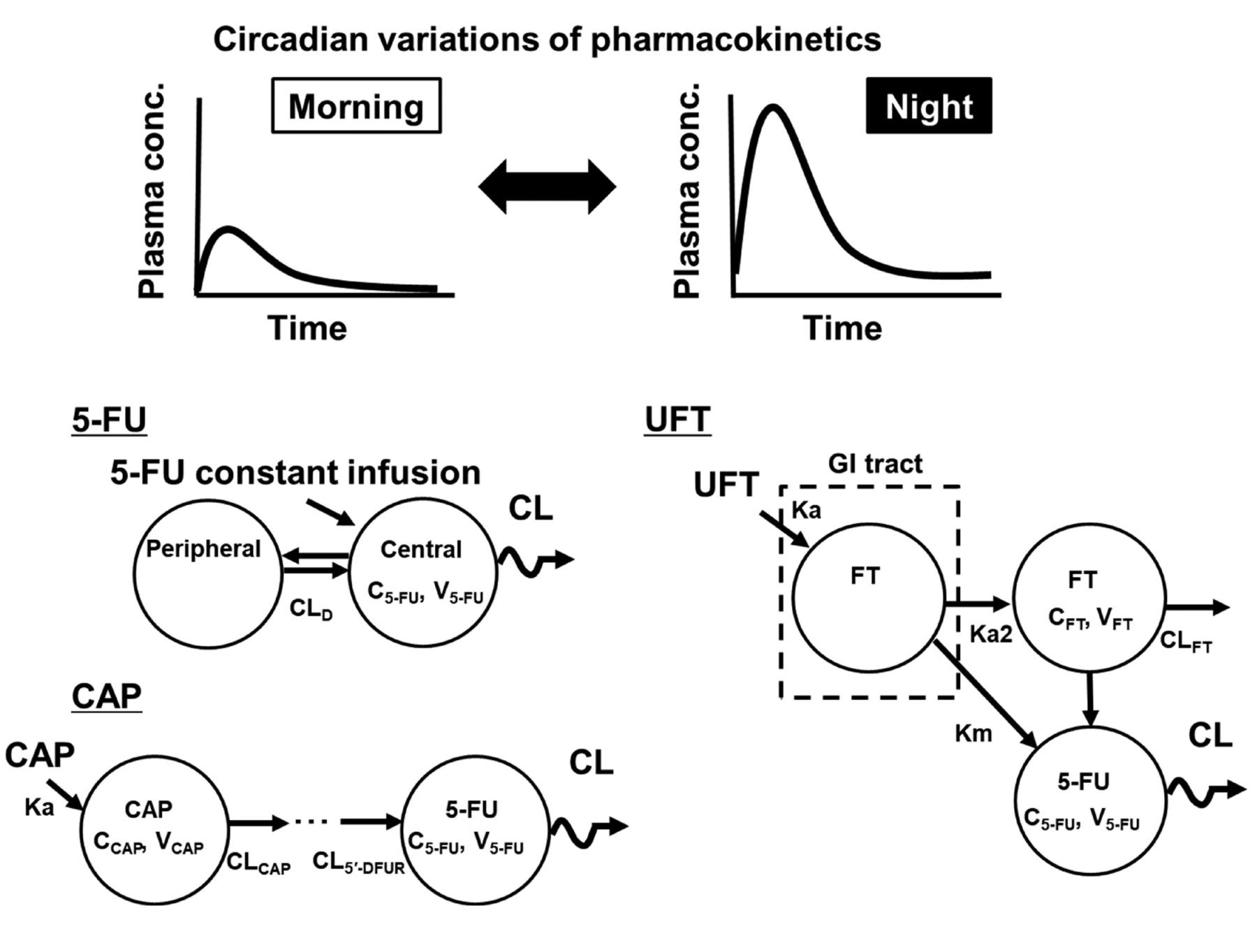
Schematic of the pharmacokinetic (PK) model taking into account the circadian rhythm of 5-fluorouracil (5-FU) after continuous infusion or of 5-FU prodrugs. To determine the circadian variations in plasma 5-FU concentrations, the cosinor method is applied in the PK model and 5-FU clearance (CL) is defined using the cosine curve. These models were developed in animal studies. CAP: Capecitabine; CCAP/FT/5-FU: plasma concentration of CAP/tegafur/5-FU; CL: clearance of 5-FU; CLCAP: CL of CAP; CLD: intercompartmental clearance; CLFT: clearance of FT; CL5’-DFUR: CL of 5’-deoxy-5-fluorouridine; FT: tegafur; GI: gastrointestinal tract; Ka: absorption rate constant; Km: conversion rate constant of FT into 5-FU during first-pass metabolism; Ka2: conversion rate constant of the first pass metabolism of FT; UFT: FT/uracil; VCAP/FT/5-FU: distribution volume of CAP/FT/5-FU.
Similar to regimens proposed for continuous 5-FU infusion, chronomodulated regimens using orally available prodrugs of 5-FU, such as capecitabine and UFT, have been proposed for obtaining favourable antitumor effects because they can help avoid 5-FU elimination (28-31). Chronomodulation can easily be achieved in such regimens involving orally available drugs via drug self-administration by patients. However, there are conflicting results regarding the utility of chronomodulated chemotherapy involving oral 5-FU prodrugs. Pilancı et al. evaluated the usefulness of capecitabine morning and noon dosing as part of a first-line XELOX regimen in patients with metastatic colorectal cancer (31). Good clinical response with favourable toxicity profiles was obtained, indicating that chronomodulation of capecitabine dosing may provide a valuable therapeutic option. In contrast, Qvortrup et al. reported that the chronomodulated XELOX regimen did not improve clinical efficacy or reduce toxicity in their patients (29). A recent phase I study showed that there were no circadian variations in exposure to capecitabine and its metabolites (5’-deoxy-5-fluorocytidine and 5’-deoxy-5-fluorouridine), including 5-FU, after continuous chronomodulated administration (32).
To investigate the usability of chronomodulated regimens involving capecitabine, circadian rhythmicity in the PK (i.e. chronopharmacokinetics) of capecitabine and its metabolites was evaluated in rats by using a population PK model with the cosinor method (25). Significant circadian variations were observed in the plasma concentration profiles of capecitabine, 5’-deoxy-5-fluorocytidine and 5’-deoxy-5-fluorouridine, and 5-FU according to the dosing time. Similar to the model for capecitabine, a population PK model taking into account the circadian rhythm showed a circadian pattern of 5-FU clearance after UFT administration to rats (26); such a circadian pattern has not been clearly obtained in clinical studies (33, 34). These animal studies using PK models with circadian rhythm showed that circadian variations in the absorption of prodrugs and their sequential metabolism to 5-FU would also contribute to variations in plasma 5-FU level. These observations suggest that the administration time point for 5-FU prodrugs is a critical factor for achieving appropriate clinical outcomes in patients. A PK model that can help determine circadian variations by the cosinor method can provide evidence to support the development of a suitable dosing strategy for improving antitumor efficacy and minimizing severe toxicities.
PK-PD/TD Model of 5-FU
PK-PD model for antitumor effects. The PK-PD model for determining tumour size profiles after treatment with antitumor agents is a valuable tool for drug development and pre-clinical and clinical studies for establishing personalized medicine. Simeoni et al. successfully developed a PK-PD model of 5-FU for tumour growth dynamics in in vivo animal studies using xenograft models (35); this model is the standard model for evaluating and comparing the degree of antitumor effects in pre-clinical drug development. In this tumour growth model, an exponential tumour growth pattern is described; it assumes that the anticancer treatment inhibits the proliferation of some cells and eventually leads to their death. Inhibition of the tumour growth rate by 5-FU is described as a factor proportional to an index of 5-FU efficacy. A transit compartment model has been applied for describing the delayed antitumor effects of 5-FU; this model allows for prediction of time delay between drug administration and the observed effects. The tumour growth model has been modified according to the cell death mechanism noted after 5-FU exposure. On the basis of Simeoni et al.’s model (35), Sung et al. proposed new PD models connected to the physiologically based pharmacokinetic model in order to describe tumour cell growth after UFT administration: The cell cycle phase– specific model, and the dual-transit compartment model where two cell death pathways exist (36). They found that the dual-transit compartment model explained the tumour growth curves in animals well, suggesting that it can be used to develop dosing strategies and patient-specific 5-FU therapies. The tumour growth model developed by Simeoni et al. is a platform for investigating responses to drug exposure in the oncology field (35); therefore, this model has been used for analysing combination chemotherapy with other antitumor agents (37) and for translational research on 5-FU (38). Daryani et al. scaled a pre-clinical PK-PD model of 5-FU to children and simulated various 5-FU dosing strategies and tumour-growth inhibition in order to determine an appropriate 5-FU dosage for use in a clinical study involving children with ependymoma (38). This translational PK-PD approach is preferable for bridging pre-clinical and clinical studies and can be applied to developing new or optimizing existing dosing strategies for 5-FU.
PK-PD model along with biomarkers for PK and PD estimation for 5-FU. The endogenous DPD substrate uracil is metabolized to dihydrouracil (UH2) in the liver. Because 5-FU is also metabolized by the same pathway, pre-therapeutic assessment of the plasma concentration ratio of UH2 to uracil was proposed as a biomarker for estimating 5-FU clearance before its administration (16). Many clinical and animal studies have shown that the pre-therapeutic UH2/uracil ratio represents a valuable indirect biomarker that shows good correlation with hepatic DPD activities (16, 39, 40), 5-FU clearance (41, 42), and 5-FU-related toxicity (43-45). On the basis of the tumour-growth model developed by Simeoni et al. (35), a PK-PD model involving the UH2/uracil ratio has been developed to analyse plasma 5-FU concentrations and tumour-growth inhibition in a rat model of 5-FU-treated colorectal cancer (46). In this model, the elimination rate constant of 5-FU was estimated using the plasma UH2/uracil ratio before 5-FU treatment, and the estimated values were applied to the PK-PD model. A combination strategy involving predictive biomarkers and model-based estimations of the drug response may aid in determining individual 5-FU dosage.
PK-TD model for myelosuppression. Severe treatment-related toxicity occurs in approximately 10-30% of 5-FU-treated patients (47). Myelosuppression is one of the most frequent dose-limiting toxicities related to this treatment (48). Predicting time-course alterations in blood cell counts after 5-FU treatment in patients helps establish the dosing schedule for each patient, thereby preventing treatment discontinuation. Mathematical PK-PD modelling can help determine the relationship between drug exposure and myelotoxicities, thereby predicting the onset and degree of myelosuppression due to 5-FU treatment. Semi-physiological PK-PD models of 5-FU for myelosuppression were developed for both rats and humans (Table II) to determine the time course of alterations in blood cell counts after 5-FU administration.
Summary of pharmacokinetic-pharmacodynamic/toxicodynamic (PK-PD/TD) model of 5-fluorouracil (5-FU).
Summary of pharmacokinetic-pharmacodynamic/toxicodynamic (PK-PD/TD) model of 5-fluorouracil (5-FU).
The Friberg model is a standard and versatile model for investigating anticancer drug–induced myelosuppression (49). The original model was developed to determine time-course alterations in white blood cell counts due to 5-FU. The model consists of three types of compartments: Proliferative cell compartment, transit compartments with maturing cells, and circulating blood cell compartment. To explain the delay in onset of myelosuppression due to 5-FU, a transit compartment model was applied to the original model, that is, the Friberg model. Another feature of the Friberg model is the explanation of regulation of the haematological system by endogenous growth factors and cytokines as a feedback mechanism. This feedback was modelled as the ratio of circulating blood cell counts at baseline divided by the cell counts at time ‘t’ raised to a feedback factor, which allows for description of the rebound of cells (overshoot compared with the baseline) after drug exposure. In clinical practice, the subsequent treatment course is generally initiated before the blood cell counts return to baseline, which makes it difficult to observe the rebound of cells after anticancer drug therapy and limits PD model development from clinical data sets. Therefore, Friberg et al. stated that this myelosuppression model should preferably be developed from animal data (49). The authors tried to extrapolate the time course of alterations in leucocyte counts from rats to those in patients by using the myelosuppression model, while accounting for the differences in drug potency in the two species (50). To determine the alterations in different blood cell counts (thrombocytes and erythrocytes), several modified models were reported for both animals and patients after these studies by Friberg et al. (Figure 2) (51-58). The Friberg model (49) provides a robust platform for analysing myelotoxicity due to chemotherapy involving a prodrug of 5-FU or their combination with other drugs (59).

Reference schematic of the pharmacokinetic-pharmacodynamic/toxicodynamic (PK-PD/TD) model of 5-fluorouracil (5-FU) for analysing the tumour volume, myelosuppression, erythropenia, and body weight loss after 5-FU treatment. These basic models have been developed using animal data. A1: Amount of 5-FU in the central compartment; A2: amount of 5-FU in the peripheral compartment; BWSS: maximal body weight; C1: 5-FU concentration in the central compartment; C2: 5-FU concentration in the peripheral compartment; Circ: circulating blood cell count; Circ0: baseline value of circulating blood cells; Edrug: drug effects; k1, first-order rate constant of transit; k12: rate constant of central compartment to peripheral compartment; k2, measure of drug potency; k21: rate constant of peripheral compartment to central compartment; kbw,in: rate constant describing the rate of body weight increase; kbw,out: first-order rate constant describing the rate of body weight decrease; kcirc: degradation rate of circulating blood cells; kin: precursor production rate; Km: concentration of 5-FU when the rate of nonlinear elimination is at half its maximum value; kprol: proliferation rate constant determining the rate of cell division; kslope: slope of linear function in drug effect; ktr: first-order rate constant of transit; V1: central volume of distribution of 5-FU; V2: peripheral volume of distribution of 5-FU; Vmax: maximal rate of saturable metabolism; XBW: one compartment of observed body weight; XCirc: one compartment of observed circulating blood cells; Xn: some transit compartments; XPre: precursor production compartment; XProl: one compartment that represented proliferative cells such as stem cells and progenitor cells; γ: power which describes a feedback mechanism from the circulating blood cells; λ0: the rate of exponential tumour growth.
In recent research involving the semi-physiological PK-PD model, a distributed delay approach was applied to model delayed responses in PK-PD studies (56, 60), instead of the traditional transit compartment model approach (49). This traditional model includes a number of different equations and has been widely accepted in describing delayed PD/TD effects, including tumour regression and myelosuppression. Although this classic model approach can adequately capture features of drug effect data, it has some disadvantages, such as the requirement for manual analysis of the preferable number of transit compartments. Moreover, many differential equations in the transit compartment model also need to fit the observed data and are not preferable for use in complex compartment models.
To overcome these disadvantages, a distributed delay approach has been proposed (56, 60). This approach utilizes an ordinary differential equation approximation of the convolution integral with gamma distribution for modelling the delay in drug absorption and the effects of drugs on myeloid cells, thereby avoiding the time-consuming process required for estimating appropriate model equations and parameters. Krzyzansk et al. (56) successfully applied the distributed delay model to previously reported myelosuppression data for FU-treated rats to which the Friberg model had been applied (49). Considering these advantages, instead of the transit compartment model, the distributed delay model should be applied as a standard model in oncology research for analysing the delays in pharmacological effects frequently observed after drug exposure in PK and PD/TD data.
Discussion and Future Perspectives
The current review discusses some modelling and simulation approaches for 5-FU that can be used to analyse drug responses after 5-FU treatment in patients; classic PK models have not been discussed. PK-PD modelling and simulation using clinical data have limitations because of the difficulties faced in collecting tumour size or drug response data from patients, whereas PK-TD model analysis uses routinely collected clinical data (i.e. blood cell counts) and is therefore relatively easy to perform. Moreover, 5-FU is administered along with other anticancer agents such as irinotecan and oxaliplatin and combination chemotherapy can also make it difficult to isolate the PD/TD data for 5-FU. To develop these types of PK-PD/TD models that can be used to analyse tumour size, drug response and combination chemotherapy data, modelling and simulation with animal data are preferable and have been reported as summarized in Table II. However, models developed using animal data cannot be directly extrapolated to clinical practice. To achieve personalized dosing strategies, approaches involving translational PK-PD/TD modelling and simulation across species and model analysis using clinical data based on the results of the animal PK-PD/TD model are required. The approach used by Daryani et al., who performed translational PK-PD modelling and simulation from a pre-clinical to paediatric model (as described in the PK-PD model for the subsection on antitumor effects), may provide a framework for future studies on the translational PK-PD/TD modelling approach for optimal dosing strategies that can reduce toxicity while maintaining the chemotherapeutic effects of 5-FU (38).
Many clinical studies have shown that there are large inter- and intra-individual variabilities in plasma 5-FU level, which contribute to clinical treatment failure (10). Contributors to inter-individual variations include differences in chemotherapeutic regimens, for example, whether bolus dosing was used, and patient characteristics. PK analysis of clinical data revealed that the area under plasma concentration-time profile (AUC: 5-FU exposure) is strongly associated with clinical outcomes, including toxicity and efficacy (11). To date, a target AUC range of 20-30 mg h l–1 in the infusion regimen has been proposed in the TDM procedure, which is simply determined from the steady-state plasma concentration and infusion period of 5-FU (11, 61). However, using this simple method for calculating AUC can lead to over- or underestimation of the values because the circadian concentration is not considered. Simulating the time profiles of both plasma 5-FU concentration and clinical responses from blood sampling data and mathematical approaches, which would enable appropriate dosing modifications for each patient, remains a critical challenge. A PK-PD/TD model that can describe the circadian rhythm of 5-FU PK may aid in realizing this strategy.
Although TDM of 5-FU can reduce toxicity and improve clinical efficacy in long-term infusion regimens, a TDM strategy has not yet been established for 5-FU prodrugs. To elucidate the relationship between exposure to the drug and its toxic properties, some studies performed PK-TD model analysis of 5-FU prodrugs (51, 57). Recently, Oyaga-Iriarte et al. successfully developed a multicompartmental PK model for capecitabine and its metabolite in patients and determined optimal sampling times for capecitabine during TDM procedures (62). These proposed sampling times will help predict the PK of capecitabine in new patients, enabling dose adjustment. Chronomodulated chemotherapy of 5-FU prodrugs has also been proposed (28-31). To determine the diurnal cycle of PK properties, some circadian models were used in animal research (25, 26). These pharmacometrics data indicate that further clinical studies on treatment efficacy and toxicity during individualized treatment with TDM need to be performed with large patient populations in order to realize the goal of personalized medicine using 5-FU prodrug chemotherapy.
Combination chemotherapy such as the FOLFIRI and FOLFOX regimens are standard approaches for treating colorectal cancer. Recently, the frequency of co-administration of drugs has increased; the folinic acid, 5-FU, irinotecan, and oxaliplatin (FOLFIRINOX) regimen has been used in chemotherapy for colorectal and pancreatic cancer (63). Moreover, supportive therapy for nausea and vomiting requires additional drugs, which complicates drug–drug interaction (64). A PK-PD/TD model that links drug exposure and drug response has been developed for 5-FU, but it has not been used for combination chemotherapy involving 5-FU (63). Developing a PK-PD/TD model for each combination chemotherapeutic regimen remains challenging.
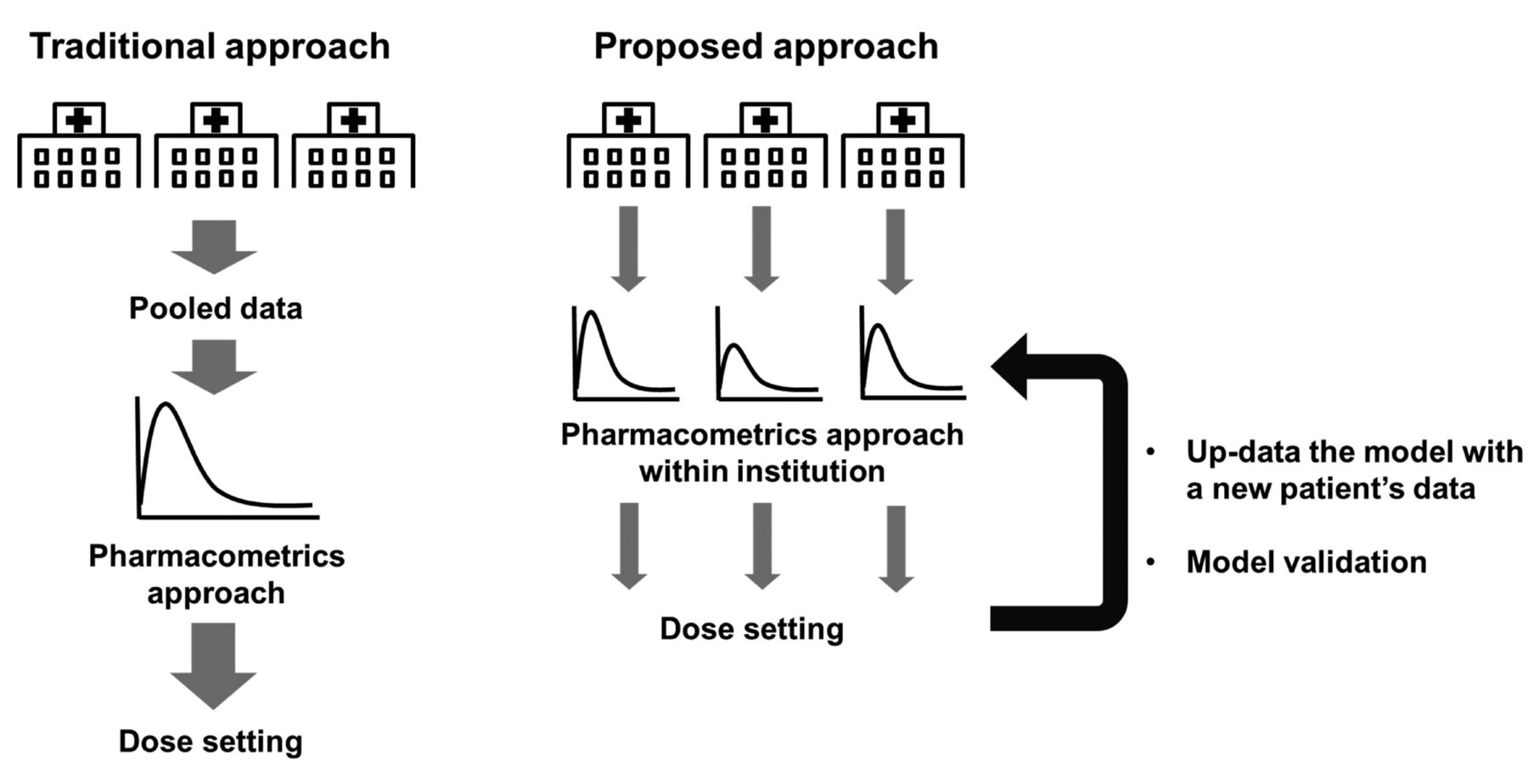
Future studies should apply the PK-PD/TD model as a tool for determining 5-FU dosing in clinical practice. Although the currently used procedure for TDM of 5-FU therapy leads to improvement in efficacies and reduction in toxicity, drug responses cannot be predicted from the plasma 5-FU level. The current dose-adjustment method requires completion of a number of cycles of therapy to achieve a narrow target therapeutic range, and the 5-FU dosage to be used in the first cycle is determined on the basis of an empirical index, namely the BSA (10, 65). PK-PD/TD modelling and simulation can facilitate more appropriate clinical dose setting for each patient via close co-operation between physicians and pharmacometricians. The traditional PK-PD/TD model has been developed using clinical data for large populations in multiple multi-institutional joint studies (Figure 3). However, there is bias with respect to patient background in the data from each clinical hospital (i.e. regionality, chronic disease, or hospitals specializing in dialysis, paediatrics, or transplantation). These different hospital characteristics may generate variabilities in the PK of 5-FU and affect dose setting. Therefore, PK-PD/TD models should be developed using routinely collected medical record data from clinical organizations. Use of a specific model for each clinical organization can help realize the goal of personalized medicine within the hospital. The model should be routinely updated with new patient information. Although the pharmacometrics technique has limitations such as model validations and education of clinical pharmacometricians, we believe that the clinical pharmacometrics approach can aid in determining the appropriate 5-FU dose to improve clinical outcomes.

{kind=link}
{kind=link}
{kind=link}
Summary of traditional and proposed prospective pharmacometrics-based approach for realizing personalized dose setting.
Conclusion
The current review promotes the understanding of the PK-PD/TD model of 5-FU. Personalized medicine involving 5-FU dose setting by using the PK-PD/TD model has not yet been applied in clinical chemotherapy. The review discusses models and current pharmacometrics approaches for personalized medicine, and provides fundamental information for further development of the PK-PD/TD model and platform. This information can help establish rational dosage-based 5-FU dose setting for each combination chemotherapy regimen, which would enable improved prognosis in patients with cancer.
Acknowledgements
The Authors would like to thank Professor Toshiyuki Sakaeda from Kyoto Pharmaceutical University (Kyoto, Japan) for his valuable support and advices during the writing of this review.
Footnotes
Authors’ Contributions
S.K.: Conception and study design, analysis, and interpretation of data, drafting of the article; Y.I.: Collection and interpretation of data, and revision of the article. All Authors approved the final version of the article.
This article is freely accessible online.
Conflicts of Interest
The Authors declare no competing interests.
- Received October 16, 2020.
- Revision received October 28, 2020.
- Accepted October 29, 2020.
- Copyright © 2020 International Institute of Anticancer Research (Dr. George J. Delinasios), All rights reserved.