

Abstract
Background: The ultraviolet A (UVA) spectrum mainly includes the region associated with the phototoxicity of fluoroquinolone antimicrobial agents. This study investigated apoptosis induced with UVA light and enoxacin in HL-60 cells. Materials and Methods: HL-60 cells were irradiated by UVA (1.1 mW/cm2) for 20 min in the presence or absence of enoxacin. The induction of apoptosis was investigated by analysing cell morphology, flow cytometry of annexin V-positive cells, DNA ladder formation, and caspase-3 activation. Results: Significant induction of apoptosis, DNA fragmentation, and caspase-3 activation were observed in cells treated with both UVA and enoxacin. UVA-induced apoptosis was significantly suppressed when NaN3, a singlet oxygen scavenger, was present. Conclusion: Apoptosis was induced by the combination of UVA and enoxacin in HL-60 cells, and singlet oxygen appears to play an important role in photodynamically-induced apoptosis.
- Apoptosis
- ultraviolet A
- enoxacin
- HL-60
- reactive oxygen species
- caspase-3
Ultraviolet (UV) light from the sun is a major cause of skin carcinogenesis (1-3). UVA (320-400 nm) is the predominant UV band and reaches the earth's surface. UVA penetrates the skin more deeply than UVB, and generates reactive oxygen species (ROS), which cause oxidative damage, in both the epidermal and dermal skin layers. Studies have shown that UVA causes ROS-mediated oxidative damage to the skin through peroxidation of lipid, and oxidative damage of DNA, resulting in the formation of 8-oxoguanine (4, 5). Previous studies have also reported that UVA causes DNA damage through photosensitization reactions in the presence of photosensitizing compound; this may induce skin cancer.
Fluoroquinolones are widely used antibiotics that play an important role in the treatment of human and animal infections (6). The absorption spectrum associated with the phototoxicity of fluoroquinolone antimicrobial agents mainly lies on the UVA region (7-10). The fluoroquinolones strongly inhibit bacterial DNA gyrase, and type II DNA topoisomerase in mammalian cells (7, 8, 11). Recently, it was also shown that fluoroquinolones can act as photosensitizers when administered to UVA-irradiated experimental animals (4).
Necrosis and apoptosis are two major processes of cell death. Necrosis is a form of traumatic cell death that is most commonly induced by severe damage that causes cytoplasmic swelling and disruption of the cell membrane, leading to release of lysosomal enzymes that commonly cause an inflammatory reaction. On the other hand, apoptosis is a highly regulated and controlled death process that act as a suicide program which removes unnecessary, aged, or damaged cells (12). Cells undergoing apoptosis experience biochemical events that lead to distinct morphological characteristics, including blebbing, cell shrinkage, chromatin condensation, and nuclear fragmentation. The cells further disassemble into membrane-enclosed vesicles termed apoptotic bodies that are taken up or phagocytosed and digested by neighbouring cells (13-15). One physical agent that can trigger the apoptotic process in cells is UV light. It directly damages DNA and leads to high ROS formation (16-18). Understanding the apoptosis-inducing properties of photosensitizing agents may be useful for the evaluation of not only photoallergenicity for preventing adverse effects, but also photosensitization for treating cancer (19-25).
Recently, we found that photochemically-active fluoroquinolones, such as lomefloxacin, can significantly induce apoptosis of human promyelocytic leukaemia cells when activated by UVA (12, 26, 27).
In this study, we investigated the ability of enoxacin for apoptosis induction, which is useful in determining the extent of drug photosensitivity (28).
Materials and Methods
Chemicals. Enoxacin, sodium azide, mannitol, and superoxide dismutase (SOD) were purchased from Sigma-Aldrich (St. Louis, MO, USA). All other reagents used were of analytical grade (2, 29).
Cell culture. Human promyelocytic leukaemia HL-60 cells were obtained from the Riken Gene Bank (Tokyo, Japan). Since leukemia cells are known to easily undergo apoptosis, HL-60 cells were used to determine the sensitivity of response in this experiment. Cells were maintained in RPMI 1640 medium containing 10% heat-inactivated foetal bovine serum (Gibco BRL, Tokyo, Japan), 100 U/ml penicillin G, 100 μg/ml streptomycin, and 2 mM glutamine (Sigma-Aldrich) in a humidified atmosphere of 5% CO2 at 37°C. When cells were in the exponential growth phase, they were used for exposure experiments. HL-60 cells were harvested and washed twice in phosphate-buffered saline (PBS; pH 7.4) before UVA exposure. The HL-60 cells were then resuspended at a density of 2×106 cells/ml in 2.5 ml of serum-free RPMI 1640, and transferred into a 35×10-mm polystyrene tissue culture dish (Corning, Corning, NY, USA) for exposure to UVA. Immediately before exposure, 200 μM enoxacin was added to the cell suspension (30).
UVA irradiation. UVA irradiation was performed using six parallel FL20SBLB lamps (Toshiba, Tokyo, Japan) with peak emission frequency of 352 nm. Cells were exposed at a distance of approximately 20 cm from the lamps. The UVA intensity was measured using a radiometer (UVR-305/365; Toshiba) that was placed at the same distance from the UVA source as the cells. The UVA irradiation dose applied throughout the 20-min exposure was 0.84 J/cm2. The culture samples treated with enoxacin alone were placed in the same position for the same period as cells were exposed to UVA. After treatment, HL-60 cells were once again incubated in a humidified atmosphere of 5% CO2 at 37°C (30, 31) and analysed as described below.
Evaluation of apoptosis and cell damage. HL-60 cells were analysed using a phase-contrast inverted microscope (Olympus, Tokyo, Japan) under 400× magnification. The integrity of the treated cells was determined by staining the cells with Trypan blue immediately after treatment. The proportion of apoptotic cells was calculated on a glass haemocytometer as the number of unstained cells showing morphological changes divided by the total number of unstained cells (30). The integrity of intact cells was also examined immediately prior to each series of treatments, with cell suspensions exhibiting >99% integrity being used in the experiments. The proportion of total sampled cells to intact cells before treatment was used as the baseline for the integrity determination (31).
Treated and control cells were incubated for 6 h, harvested and collected by centrifugation at 300 × g for 5 min at room temperature. Phosphatidylserine externalization was then evaluated by staining with Muse Annexin V and Dead Cell Assay Reagent (EMD Millipore Bioscience, Burlington, MA, USA) according to the manufacturer's instructions. Cells staining positively with 7-amino-actinomycin D (7-AAD), annexin V-fluorescein isothiocyanate, or both were quantified by flow cytometry using a Muse Cell Analyzer (EMD Millipore Bioscience). The apoptotic ratio was determined from the four identified cell populations: cells not undergoing detectable apoptosis (annexin V-negative and 7-AAD-negative cells); early apoptotic cells (annexin V-positive and 7-AAD-negative cells); late apoptotic cells (annexin V-positive and 7-AAD-positive cells); and necrotic cells (annexin V-negative and 7-AAD-positive cells (32).
Electrophoretic analysis for DNA fragmentation. The cells were harvested, washed in PBS (pH 7.4), and lysed in 100 μl of 0.1 M phosphate-citrate buffer. Following lysis, the samples were centrifuged at 16,000 × g for 5 min. The supernatants were then treated with 200 μg/ml DNase-free RNase at 37°C for 1 h, followed by 1 μg/ml proteinase K at 50°C for 1 h. The samples were separated by electrophoresis on 1.5% (w/v) agarose gels containing 1 μg/ml ethidium bromide. The DNA fragments (DNA ladders) were visualized using a UV transilluminator (ATTO, Tokyo, Japan). The sizes of the DNA fragments were determined by comparison to DNA molecular weight markers (100-bp DNA ladder; Invitrogen, Carlsbad, CA, USA) (29, 30).
Measurement of caspase-3 activity. Caspase-3 activity was assayed using the specific fluorogenic substrate, N-acetyl-Asp-Glu-Val-Asp-7-amino-4-trifluoromethylcoumarin (Ac-DEVD-AFC; MBL, Tokyo, Japan). After incubation, treated cells were washed with 50 mM PBS (pH 7.4) and resuspended in buffer containing 50 mM Tris-HCI (pH 7.4), 1 mM ethylenediaminetetra-acetic acid, and 10 mM ethylene glycol-bis(β-aminoethyl ether)-N,N,N’,N’-tetra-acetic acid (33). The cell lysate was then centrifuged at 800 × g for 5 min, and the supernatant was incubated with peptide substrate (50 μM) at 37°C for 2 h. The formation of 7-amino-4-trifluoromethylcoumarin was measured using a fluorescence spectrophotometer (F-3000; Hitachi, Tokyo, Japan) with an excitation wavelength of 400 nm and an emission wavelength of 505 nm. The enzymatic activity measured immediately prior to each experiment was used as the control value. The caspase activity was expressed as the ratio of released 7-amino-4-trifluoromethylcoumarin under the experimental condition to that of the untreated control (30).
Effect of ROS scavengers. To determine whether ROS, including singlet oxygen, superoxide radicals and superoxide radicals, participate in the induction of apoptosis due to UVA combined with enoxacin, we examined the effect of ROS scavengers (10 mM NaN3, 100 mM mannitol or 100 μg/ml SOD) on the proportion of cells exhibiting phosphatidylserine externalization by annexin V staining (30).
Statistical analysis. The results are expressed as the mean±standard deviation (S.D.). The values were compared by one-way analysis of variance, with p=0.05 as the minimum level of significance (34).
Results
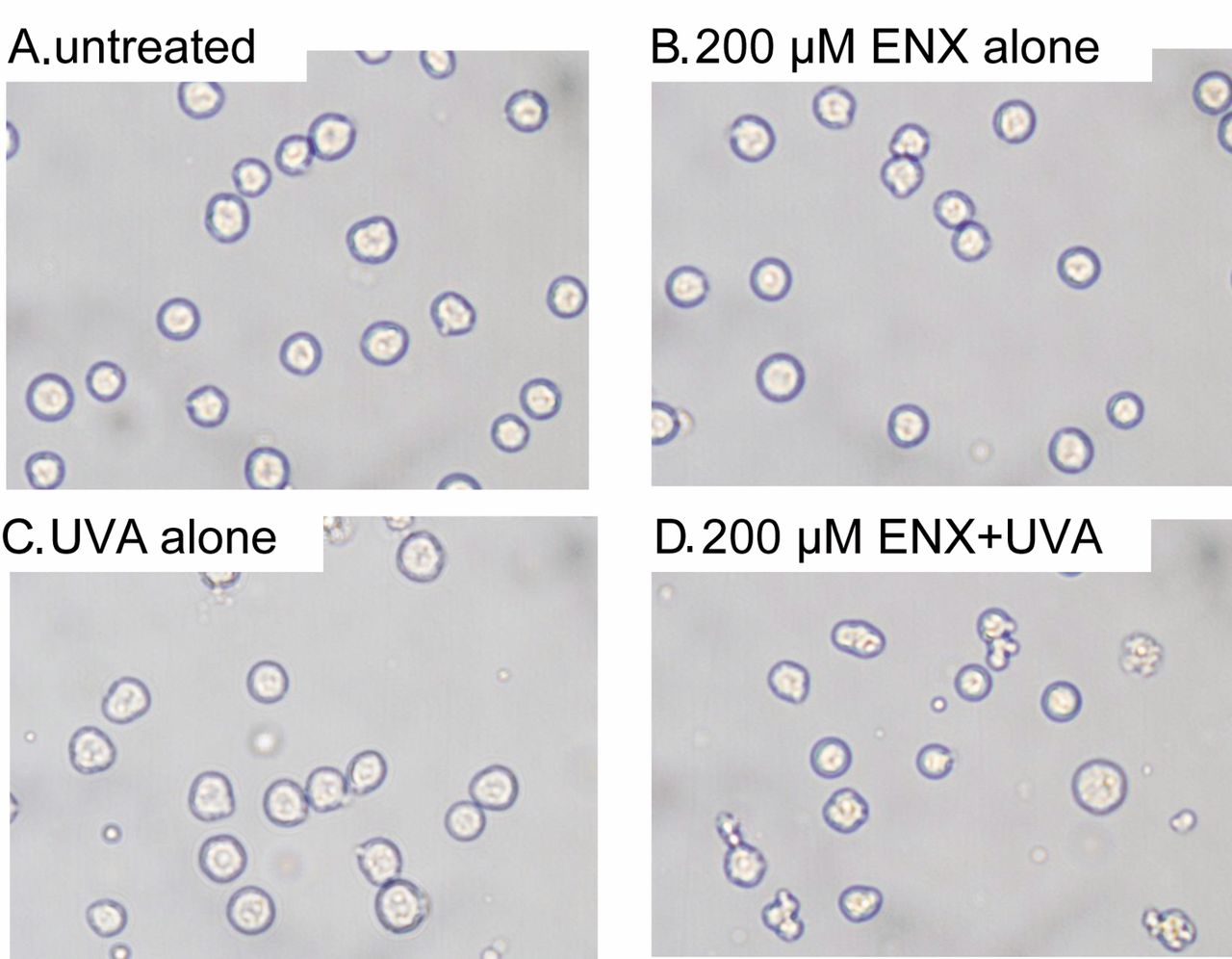
Morphological changes. Apoptosis was studied by examining the morphological changes of cells using phase-contrast microscopy after a 4-h incubation period following the treatment with enoxacin alone, UVA alone, or enoxacin plus UVA (Figure 1). There were no significant morphological changes in cells treated with enoxacin alone (Figure 1B) or UVA alone (Figure 1C). In contrast, membrane blebbing and cell shrinkage were clearly observed in cells treated with enoxacin plus UVA (Figure 1D).
Induction of apoptosis. Induction of apoptosis and necrosis was measured by flow cytometry 6 h after treatment. Figure 2 shows representative dot charts of annexin V/7-AAD bivariate flow cytometry after a 4-h incubation period following treatment with enoxacin alone, UVA alone, or enoxacin plus UVA, or no treatment. The dot-plot histogram revealed that there was no significant induction of apoptosis in cells treated with enoxacin alone (Figure 2B) or UVA alone (Figure 2C). In contrast, early apoptosis, i.e. cells positive only for annexin V, was clearly seen in the enoxacin plus UVA-treated cells (Figure 2D).

Analysis of cellular morphology using phase-contrast microscopy 4 h after no treatment (A), and treatment with 200 μM enoxacin alone (B), UVA alone (C), or 200 μM enoxacin + UVA (D).
Figure 3 shows the proportion of early apoptotic HL-60 cells over the duration of the incubation period after treatment. Under all conditions, the proportion of early apoptotic cells was less than 2% immediately following the initiation of the treatment. In cells treated with enoxacin plus UVA, there was a significant increase in the proportion of early apoptotic cells over time. HL-60 cells that were treated with enoxacin at 200 μM underwent apoptosis. The proportion of early apoptotic cells reached a maximum after 4 h, and subsequently decreased. No significant increase in the proportion of apoptotic cells was observed in cells treated with UVA or enoxacin alone.
DNA fragmentation. To further explore the mechanism of UVA-mediated apoptosis with enoxacin in these cells, agarose gel electrophoresis was performed using DNA samples from the HL-60 cells. No distinct DNA ladder was observed for cells treated with enoxacin or UVA alone (Figure 4, lanes 3 and 4, respectively). In contrast, a DNA ladder was clearly observed 4 h after exposure to UVA for cells treated with 200 μM enoxacin with UVA (Figure 4, lanes 5 and 6, respectively).
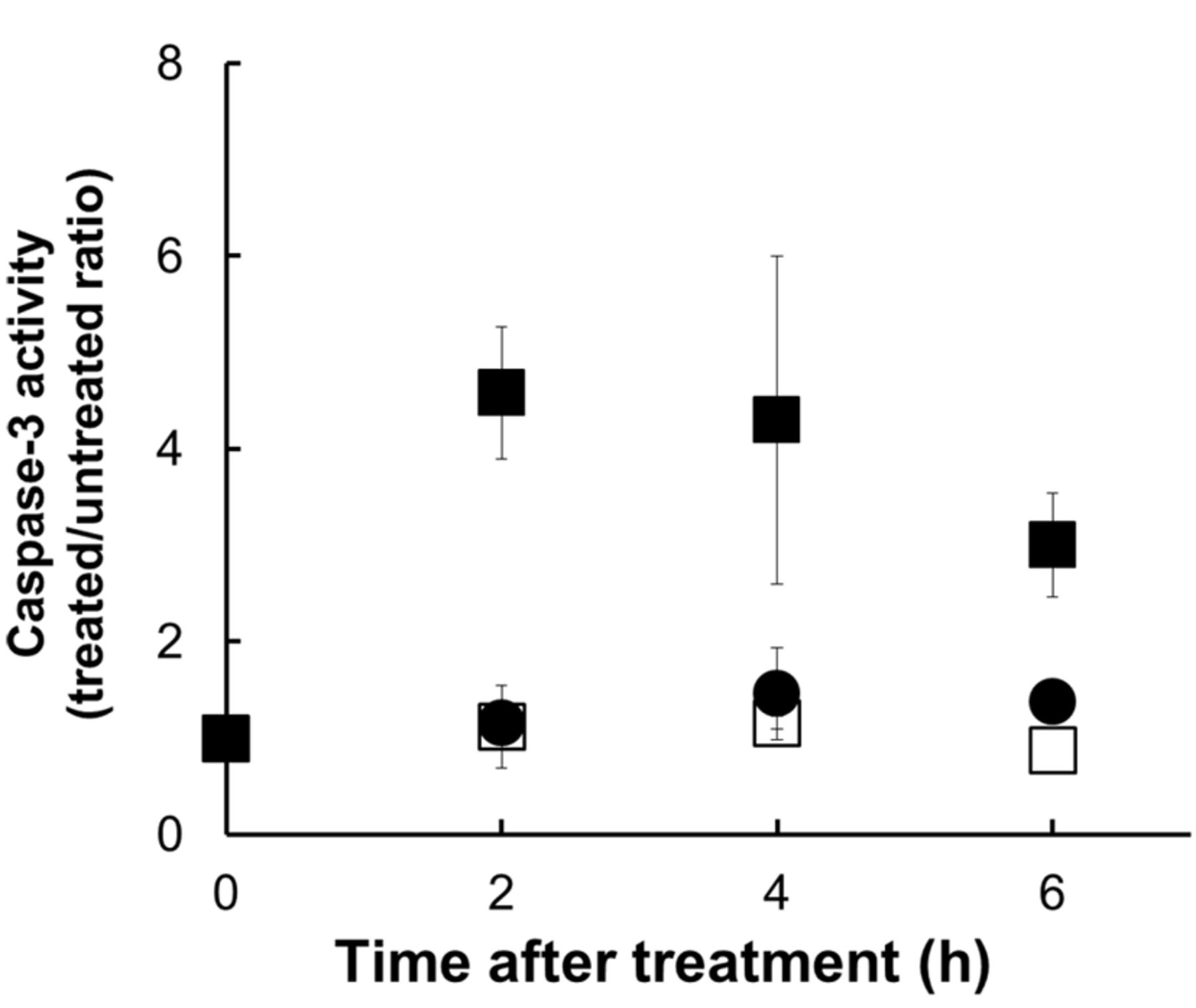
Caspase-3 activation. To investigate whether the activation of caspases by UVA and enoxacin lead to apoptosis of HL-60 cells, the enzymatic activity of caspase-3 was measured using a peptide substrate which becomes fluorescent when cleaved by active caspase-3 (Figure 5). We observed that caspase-3 activity increased, peaking at 1 h after treatment, in cells treated with UVA in the presence of 200 μM enoxacin. No increase in caspase-3 activity was observed in cells treated with UVA or enoxacin alone.
Effect of ROS scavengers. To elucidate the mechanism of apoptosis induced photodynamically, we next investigated the effect of ROS scavengers on the induction of apoptosis with UVA with/without enoxacin (Figure 6). NaN3 significantly suppressed the induction of apoptosis in cells treated with enoxacin plus UVA. In contrast, SOD and mannitol did not appear to affect the induction of apoptosis (12, 30).
Discussion
Firstly, we examined whether exposure to UVA in the presence of enoxacin induces apoptosis in HL-60 cells. We found that UVA-induced apoptosis was greatly enhanced by enoxacin, morphological indicators such as membrane blebbing, cell shrinkage, formation of membrane protrusions and breaking of cells into apoptotic bodies were clearly observed in cells treated with both UVA and enoxacin, while no significant morphological changes were observed in cells exposed to either UVA or enoxacin alone (30). The proportion of apoptotic cells among enoxacin plus UVA-treated cells increased by more than one order of magnitude when compared to those treated with enoxacin or UVA alone. These results clearly showed the synergistic effect of enoxacin and UVA on the induction of apoptosis (30).
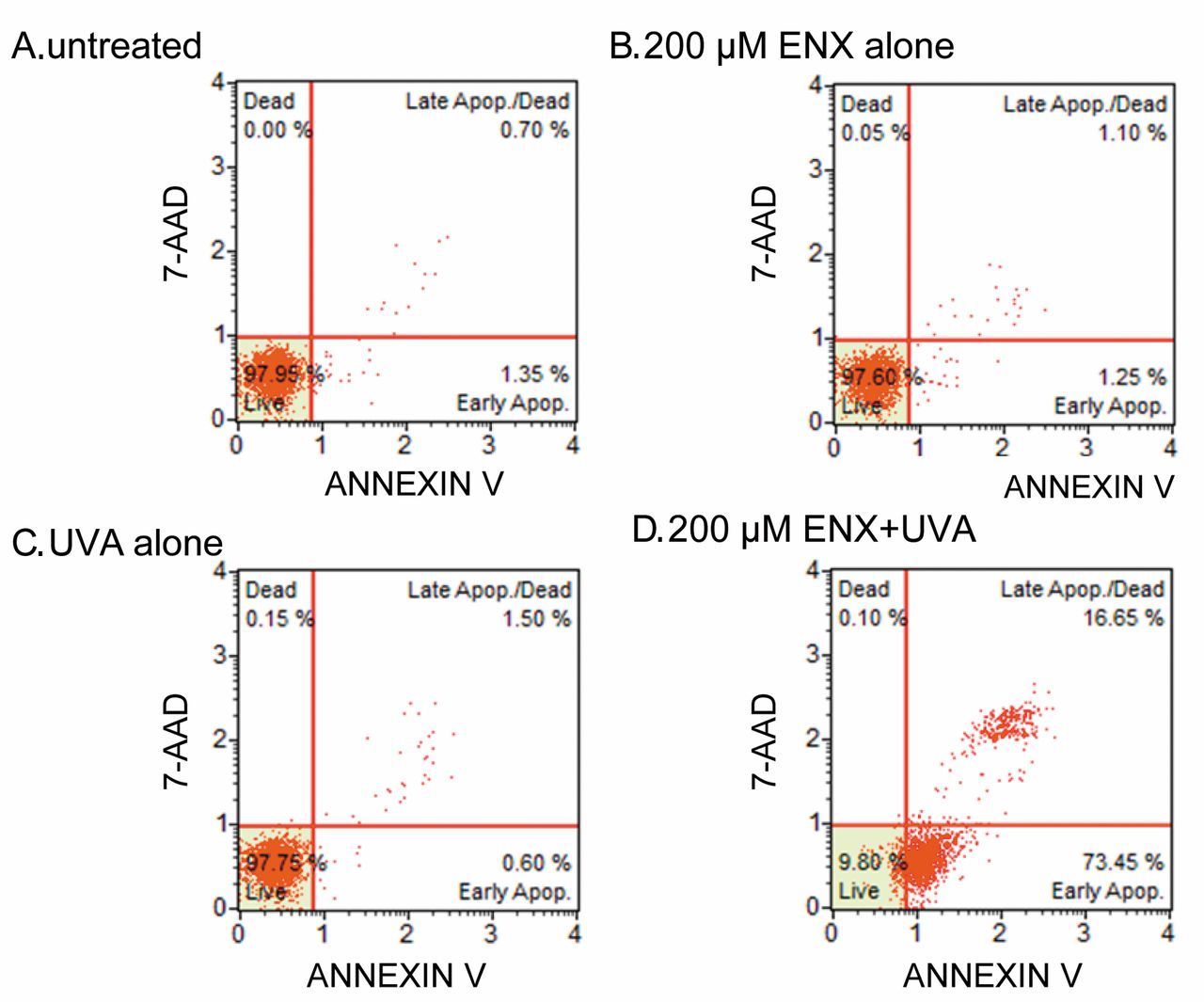
Representative dot charts of annexin V/7-amino-actinomycin D bivariate flow cytometry after 4-h incubation of HL-60 cells with no treatment (A), and treatment with 200 μM enoxacin alone (B), UVA alone (C), or 200 μM enoxacin + UVA (D).
The fragmentation of DNA at linker regions between nucleosomes into fragments of multiples of 180-200 base pairs in length is reported to be a hallmark of apoptosis (31-33). In this study, enoxacin plus UVA treatment resulted in the formation of a characteristic DNA ladder on agarose gel electrophoresis. This was not observed immediately after exposure (data not shown), but was clearly observed 4 h later, suggesting that the DNA fragmentation was caused by an enzymatic process rather than by photodynamic activation of enoxacin (30, 32, 34).

Proportion of apoptotic HL-60 cells following UVA exposure in the presence or absence of 200 μM enoxacin. ○ Untreated; ● 200 μM enoxacin alone; □ UVA alone; ▪ 200 μM enoxacin + UVA. Values represent the mean±S.D. of three independent experiments. *Significantly different at p<0.05 in comparison to the untreated controls.
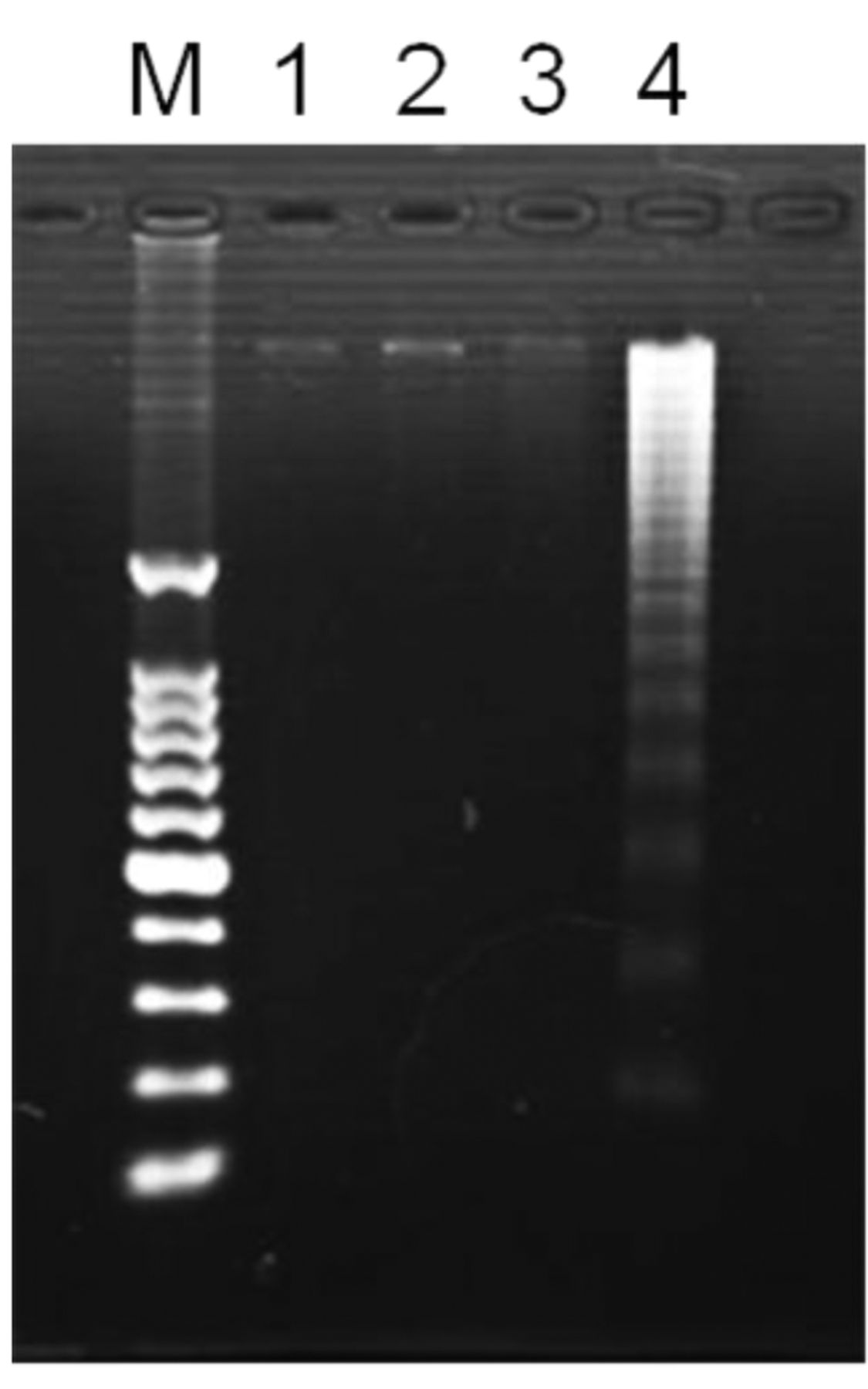
DNA ladder formation in HL-60 cells 4 h after treatment with enoxacin with and without UVA. Lane 1, DNA molecular size markers; lane 2, untreated; lane 3, 200 μM enoxacin alone; lane 4, UVA alone; lane 5, 200 μM enoxacin + UVA.
There are two kinds of caspases, initiator caspases and effector caspases. Caspase-3 which is an effector caspase, is activated by active initiator caspases via proteolytic cleavage. Caspase-3 plays an important role on the execution of the final phase of apoptosis, and it is activated in cells to carry out apoptotic death (34-36). We observed significant activation of caspase-3 after treatment with enoxacin plus UVA. Both the proportion of apoptotic cells and the activity of caspase-3 gradually increased, reaching a peak 4 h after treatment, before subsequently decreasing, which suggests that caspase-3 acted as the executor caspase responsible for the induction of apoptosis following the combination treatment. However, the mechanism by which caspase-3 is activated by treatment with enoxacin plus UVA remains to be determined. Some structure–activity relationship studies have been performed for quinolone-induced phototoxicity (4, 5), and the results have suggested that the phototoxicity of quinolones is determined by the nature of its 8-position substituent; halogens caused the greatest photoreactive activity, while hydrogen and methoxy substituents had little effect (34, 35). In the pathogenesis of various skin diseases, a number of signalling pathways are activated as a result of UVA-mediated ROS generation. ROS are believed to activate proliferative and cell survival signalling that can alter the apoptotic pathways involved in the pathogenesis of a number of skin disorders, including photosensitivity (30-36).
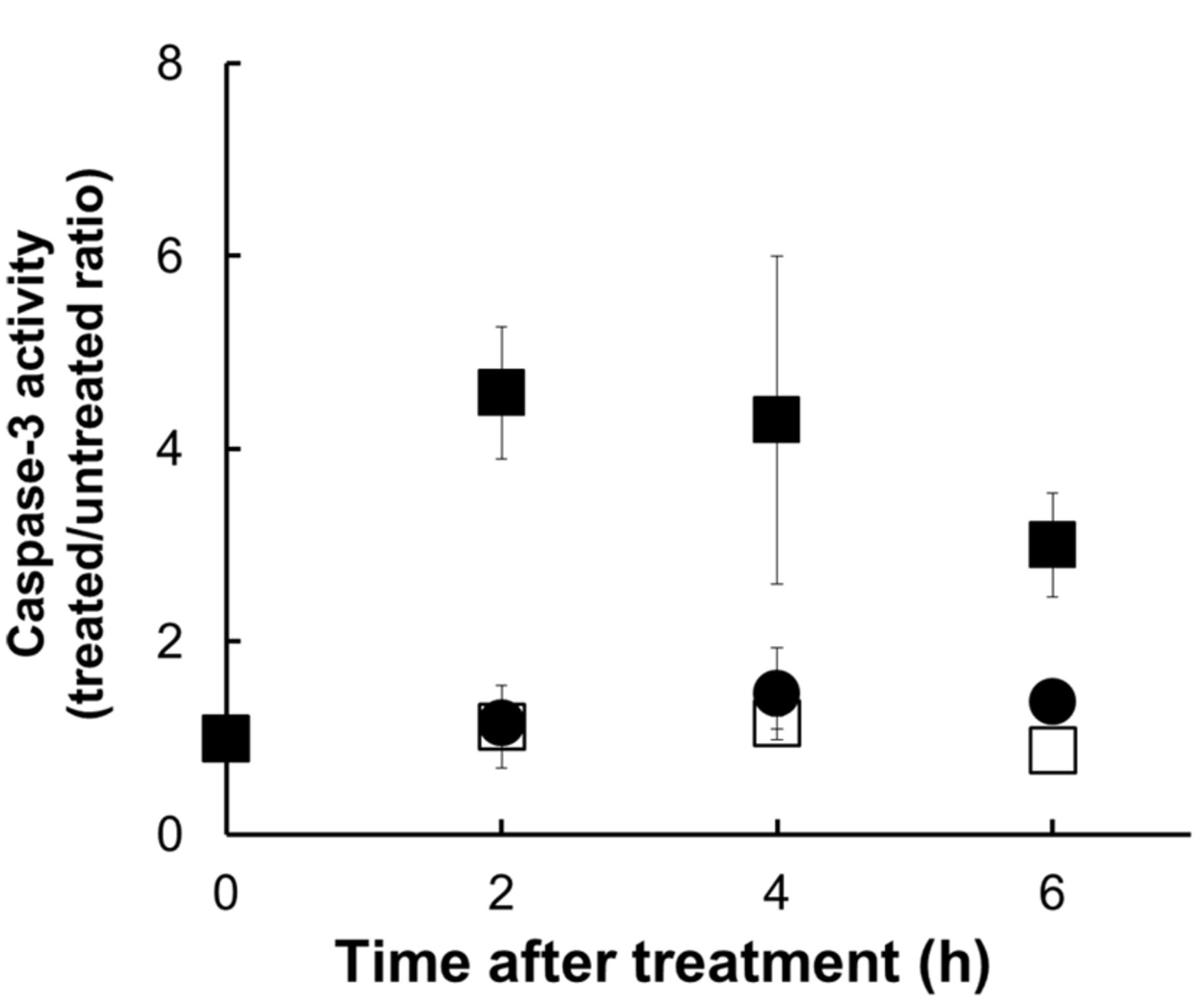
Caspase-3 activity in HL-60 cells following UVA exposure in the presence and absence of 200 μM enoxacin. □ 200 μM Enoxacin alone; ● UVA alone; ▪ 200 μM enoxacin + UVA. Values represent the mean±S.D. of three independent experiments. *Significantly different at p<0.05 in comparison to untreated controls.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Effect of reactive oxygen scavengers on UVA-induced apoptosis 4 h after treatment in the absence (○) and presence (●) of 200 μM enoxacin. Values represent the mean±S.D. of three independent experiments. *Significantly different at p<0.05 in comparison to the control cells with no scavenger (None).
The ROS scavengers used in this study included NaN3, which is reported to be scavenger of singlet oxygen; mannitol at a concentration (100 mM) that should scavenge photodynamically-induced hydroxyl radicals; and SOD, which catalyses the elimination of superoxide radicals (12, 37-38). NaN3 caused significant suppression of UVA-induced apoptosis, suggesting that singlet oxygen plays a more important role than hydroxyl radicals or superoxide in the induction of apoptosis based on the combination of UVA and enoxacin. Several mechanisms involving the generation of ROS have been proposed to explain the photodynamic action of enoxacin (22, 23). Activated high-energy enoxacin species are formed during the intermolecular interaction between excited enoxacin and oxygen molecules in the ground state, thereby resulting in intermolecular energy transfer and the formation of singlet oxygen, as follows:
 The above sensitization mechanism that leads to generation of singlet oxygen is defined as a type II photoreaction (32). In the present study, a scavenger of singlet oxygen led to significant suppression of the induction of apoptosis, while scavengers of superoxide anion and hydroxyl radical did not. Our results support a previous study that suggests that singlet oxygen stimulates apoptotic cell death signalling pathways. This knowledge may lead to the establishment of new approaches for cancer therapy that utilize the formation of singlet oxygen through UVA-mediated photosensitization of enoxacin specifically targeting tumour cells (30, 31, 37-52).
The above sensitization mechanism that leads to generation of singlet oxygen is defined as a type II photoreaction (32). In the present study, a scavenger of singlet oxygen led to significant suppression of the induction of apoptosis, while scavengers of superoxide anion and hydroxyl radical did not. Our results support a previous study that suggests that singlet oxygen stimulates apoptotic cell death signalling pathways. This knowledge may lead to the establishment of new approaches for cancer therapy that utilize the formation of singlet oxygen through UVA-mediated photosensitization of enoxacin specifically targeting tumour cells (30, 31, 37-52).
- Received December 3, 2018.
- Revision received January 17, 2019.
- Accepted January 22, 2019.
- Copyright© 2019, International Institute of Anticancer Research (Dr. George J. Delinasios), All rights reserved