

Abstract
Background/Aim: The identification of a series of oxadiazole-based compounds, as promising antiproliferative agents, has been previously reported. The aim of this study was to explore the SAR of newly-synthesized oxadiazole derivatives and identify their molecular targets. Materials and Methods: A small library of 1,2,5-oxadiazole derivatives was synthetized and their antiproliferative activity was tested by the MTT assay. Their interaction with topoisomerase I was evaluated and a molecular docking study was performed. Results: Several candidates showed cytotoxicity towards two human tumor cell lines, HCT-116 (colorectal carcinoma) and HeLa (cervix adenocarcinoma). Some derivatives exhibited inhibitory effects on the catalytic activity of topoisomerase I and this effect was supported by docking studies. Conclusion: The enzyme inhibition results, although not directly related to cytotoxicity, suggest that a properly modified 1,2,5 oxadiazole scaffold could be considered for the development of new anti-topoisomerase agents.
- Oxadiazoles
- cytotoxicity
- topoisomerase I
- HCT-116
- HeLa
- docking studies
Cancer is one of the main causes of morbidity and mortality worldwide, causing approximately 1 in 6 deaths (1).
Antiproliferative agents are most efficient at killing cells that are rapidly dividing: as such, they are usually very toxic, since they also target normal proliferating cells. Besides the adverse side-effects, the intrinsic or acquired resistance to antiproliferative drugs (2) compromises the success of the therapeutic treatments. Therefore, although antiproliferative agents play a crucial role in the reduction of mortality, the poor selectivity and the development of resistance to their action have prompted continuous efforts to discover new compounds endowed with good efficacy and reduced toxicity.
With the aim of identifying novel potential chemotherapeutic agents (3-9), we synthesized a small library of 1,2,5-oxadiazole derivatives (3) (Figure 1) related to MD77, previously disclosed by our research group (4), as an interesting anti-proliferative hit. Some derivatives had already been tested on HCT-116 cell line and showed a significant activity. Therefore, to complete our studies on this interesting scaffold, several new derivatives (Figure 2) were designed and synthesized, following a synthetic procedure reported in literature (3). Their cytotoxicity towards the HCT-116 cell line, evaluated by MTT assay, indicated that some members of the library exert growth inhibition properties. Therefore, all the derivatives (1-29) were tested on a second human cancer cell line (HeLa). In order to gain insight into their possible molecular target(s), their effects on the activity of topoisomerases (topos) I and II were examined, since several oxadiazole derivatives have been found to inhibit these enzymes (10, 11). Docking studies provided evidence of interactions of representative derivatives with topo I.
Materials and Methods
Chemistry. Reagents and solvents were purchased from Sigma-Aldrich (St. Louis, MO, USA) and used without further purification. Some reactions involving air-sensitive reagents were performed under nitrogen atmosphere and anhydrous solvents were used when necessary.

Small library of MD77 derivatives.
Reactions were monitored by thin layer chromatography analysis on aluminum-backed Silica Gel plates (pore size 60 Å, 70-230 mesh; Sigma-Aldrich), using an ultraviolet fluorescent lamp at 254 nm and 365 nm. Visualization was aided by opportune staining reagents.
Purification of intermediates and the final compounds was performed by flash chromatography using Geduran® Si 60 (40-63 μm; Merck, Darmstadt, Germany).
Melting points were determined on a Buchi Melting Point B540 instrument (Buchi, Flawil, Switzerland). 1H-, 13C- and 19F-NMR spectra were recorded at room temperature on a Varian 300 MHz Oxford instrument (Varian, Palo Alto, CA, USA), using TMS as internal standard. CDCl3, CD3OD, acetone-d6 and DMSO-d6 were used as deuterated solvents for all the spectra run. Chemical shifts are expressed as δ (ppm). Multiplicity is reported as s (singlet), br s (broadened singlet), d (doublet), t (triplet), q (quartet), m (multiplet), dd (doublet of doublets), dt (doublet of triplets). The coupling constants (J-values) are expressed in Hertz (Hz). All spectroscopic data match the assigned structures. The purity of the compounds was assessed by means of high-performance liquid chromatography, using a Varian LC-940 HPLC system (Varian, Palo Alto, CA, USA), and was ≥95% unless otherwise stated. Mass spectrometry analyses were carried out on an LTQ Orbitrap XL mass spectrometer (Thermo Fisher Scientific, Waltham, MA, USA), equipped with a Finnigan IonMax Electrospray interface.
Synthesis of 1,2,5-oxadiazol-3-amines (30a-c). The key intermediates 30a,b and the new amine 30c were obtained as reported in literature (3).
4-(1,1’-biphenyl-4-yl)-1,2,5-oxadiazol-3-amine (30c). Starting compound: 1,1’-biphenyl-4-carbaldehyde (15 mmol). Yield: 19%. Light yellow solid. M.p. 161.4-161.8°C. 1H-NMR (CDCl3): δ 4.24 (br s, 2H, NH2 exchanged with D2O), 7.38-7.52 (m, 3H, ArH), 7.62-7.66 (m, 2H, ArH) 7.74-7.84 (m, 4H, ArH) ppm. 13C-NMR (DMSO-d6): δ 125.25, 127.46, 128.00, 128.77, 128.99, 129.79, 139.79, 142.55, 147.31, 156.09 ppm. MS (ESI): calcd. for C14H11N3O 237, found 238 [M+H]+.
General procedure for the synthesis of N-(4-phenyl-1,2,5-oxadiazol-3-yl) substituted amides (15, 16 and 26). In a two-necked flame-dried flask, 60% NaH in mineral oil (0.3 mmol) was suspended in dry N-N-dimethylformamide (3 ml) under nitrogen atmosphere. The suspension was cooled in an ice bath and the appropriate key intermediate (0.25 mmol) was added. The mixture was stirred at 0°C for 20 min. Then, the suitable commercially available acyl chloride (0.3 mmol) was added dropwise at 0°C. The mixture was stirred at 60°C for 12 h. The reaction was quenched with water (3 ml), and N-N-dimethylformamide was removed under vacuum. The residue was extracted with ethyl acetate (3×2 ml); the organic layer was dried over Na2SO4 and evaporated in vacuo. The crude product was purified by column chromatography to obtain the desired adduct. Amides 21, 2 and 31 were synthesized as previously reported (2).
N-(4-(4-chlorophenyl)-1,2,5-oxadiazol-3-yl)heptanamide (15). Starting compounds: 30a and n-heptanoyl chloride. Purified by flash chromatography (eluent mixture: cyclohexane/ethyl acetate, 85:15). Yield: 43%. White solid. M.p. 115.5-115.8°C. 1H-NMR (CDCl3): δ 0.89 (t, J=6.9 Hz, 3H, CH3), 1.25-1.29 (m, 6H, CH2), 1.62-1.72 (m, 2H, CH2), 2.48 (t, J=6.9 Hz, 2H, CH2), 7.47 (d, J=8.7 Hz, 2H, ArH), 7.59 (d, J=8.7 Hz, 2H, ArH), 7,65 (br s, 1H, NH exchanged with D2O) ppm. 13C-NMR (CDCl3): δ 13.64, 22.52, 24.94, 29.95, 31.60, 35.71, 125.00, 129.31, 129.68, 136.28, 149.94, 150.63, 172.27 ppm. MS (ESI): calcd. for C15H18ClN3O2 308, found 309 [M+H]+.
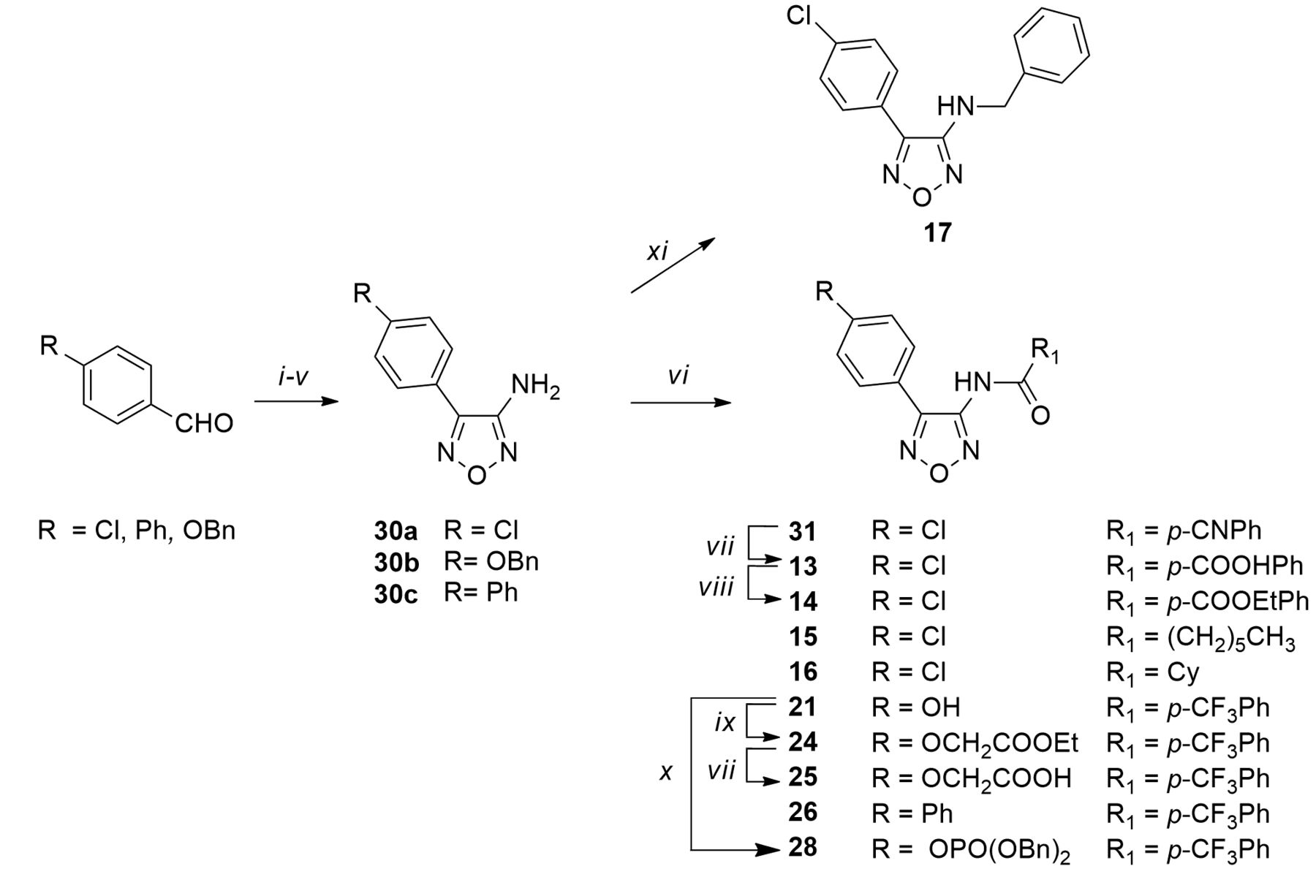
Reagents and conditions: (i) NH2OH•HCl, NaHCO3, methanol, reflux, 2 h; (ii) NCS, N,N-dimethylformamide, rt, 12 h; (iii) KCN, diethyl ether/H2O, rt, 5h; (iv) NH2OH•HCl, NaHCO3, methanol, reflux, 12 h; (v) 2N NaOH, reflux, 12 h; (vi) R1COCl, 60% NaH, dry N,N-dimethylformamide, N2, 60°C, 12 h; (vii) 1N NaOH, ethanol, reflux, 3 h for 13 or tetrahydrofuran/ethanol, reflux, 1 h for 25; (viii) (CH3CH2O)2CO, dry CH3CH2OH, 40°C, 12 h; (ix) ethyl chloroacetate, dry N,N-dimethylformamide, N2, rt, 24 h; (x) dibenzyl phosphite, 4-dimethylaminopyridine, N,N-diisopropylethylamine, acetonitrile, N2, −10°C, 3 h; (xi) a. PhCHO, CH3COOH, rt, 3 h, b. NaBH4, 0°C, 1 h.
N-(4-(4-chlorophenyl)-1,2,5-oxadiazol-3-yl)cyclohexanecarboxamide (16). Starting compounds: 30a and cyclohexanecarbonyl chloride. Purified by flash chromatography (eluent mixture: cyclohexane/ethyl acetate, 90:10). Yield: 61%. White solid. M.p. 167.5-115.9°C. 1H-NMR (CDCl3): δ 1.18-1.92 (m, 10 H, CH, CH2), 7.29 (br s, 1H, NH exchanged with D2O), 7.41 (d, J=8.4 Hz, 2H, ArH), 7.51 (d, J=8.4 Hz, 2H, ArH) ppm. 13C-NMR (CDCl3): δ 25.58, 29.39, 29.92, 45.24, 124.07, 129.16, 129.73, 137.31, 148.79, 149.49, 174.81 ppm. MS (ESI): calcd. for C15H16ClN3O2 306, found 307 [M+H]+.
4-(trifluoromethyl)-N-(4-([1,1’-biphenyl]-4-yl)-1,2,5-oxadiazol-3-yl)benzamide (26). Starting compounds: 30c and 4-trifluoromethyl-benzoyl chloride. Purified by flash chromatography (eluent mixture: cyclohexane/ethyl acetate, 80:20). Yield: 14%. White foam. 1H-NMR (acetone-d6): δ 7.37-7.50 (m, 3H, ArH), 7.70 (d, J=8.7 Hz, 2H, ArH), 7.82 (d, J=8.4 Hz, 2H, ArH), 7.90-7.97 (m, 4H, ArH), 8.30 (d, J=8.1 Hz, 2H, ArH), 10.59 (br s, 1H, NH exchanged with D2O) ppm. 13C-NMR (CDCl3): δ 124.63, 125.75 (q), 126.90, 127.45, 128.02, 128.09, 128.24, 128.93, 128.98, 133.21, 136.29, 139.67, 143.13, 149.79, 151.31, 165.27 ppm. 19F-NMR (CDCl3): δ -63.58 (s, CF3) ppm. MS (ESI): calcd. for C22H14F3N3O2 409, found 410 [M+H]+.
Synthesis of N-benzyl-4-(4-chlorophenyl)-1,2,5-oxadiazol-3-amine (17). Benzaldehyde (0.306 mmol) was added to 30a (0.153 mmol) in acetic acid (4 ml) and the solution was stirred at room temperature for 3 h. The reaction was cooled at 0°C and NaBH4 (0.306 mmol) was added; the resulting mixture was stirred for 1 h. Afterwards, a solution of 2N NaOH was added, and the mixture was extracted with ethyl acetate (3×2 ml), dried over Na2SO4, filtered and concentrated in vacuo. Compound 17 was purified by flash chromatography (eluent mixture: cyclohexane/ethyl acetate, 95:5). Yield: 67%. Brown solid. M.p. 147.5-147.7 °C. 1H-NMR (CDCl3): δ 4.20 (br s, 1H, NH exchanged with D2O), 4.52 (s, 1H, CH2), 4.58 (s, 1H, CH2), 7.25-7.45 (m, 5H, ArH), 7.38 (d, J=8.6 Hz, 2H, ArH), 7.65 (d, J=8.6 Hz, 2H, ArH) ppm. 13C-NMR (acetone-d6): δ 48.47, 125.03, 127.34, 128.02, 128.50, 129.54, 129.93, 136.06, 138.95, 146.08, 155.99 ppm. MS (ESI): calcd. for C15H12ClN3O 286, found 287 [M+H]+.
Synthesis of ethyl 4-((4-(4-chlorophenyl)-1,2,5-oxadiazol-3-yl)carbamoyl)benzoate (14). Compound 31 (0.087 mmol), diethylcarbonate (0.087 mmol) and dry MgCl2 (0.0087 mmol) in ethanol (3 ml) were stirred under argon atmosphere, at 40°C for 12 h under not conventional (12) condition. Then, the reaction mixture was diluted with water (2 ml), concentrated under reduced pressure and extracted with ethyl acetate (3×2 ml). The organic phase was dried over Na2SO4, filtered and concentrated under vacuum. Compound 14 was purified by flash chromatography (eluent mixture: petroleum ether/ ethyl acetate, 80:20). Yield: 15%. White foam. 1H-NMR (CDCl3): δ 1.43 (t, J=7.2 Hz, 3H, CH3), 4.43 (q, J=7.2 Hz, 2H, CH2), 7.47 (d, J=8.1 Hz, 2H, ArH), 7.65 (d, J=8.1 Hz, 2H, ArH), 7.94 (d, J=8.1 Hz, 2H, ArH), 8.18 (d, J=8.1 Hz, 2H, ArH), 8.31 (br s, 1H, NH exchanged with D2O) ppm. 13C-NMR (CDCl3): δ 14.47, 61.93, 124.12, 127.86, 129.07, 129.85, 130.48, 135.01, 135.57, 137.44, 148.85, 149.82, 164.93, 165.56 ppm. MS (ESI): calcd. for C18H14ClN3O4 372, found 373 [M+H]+.
Syntheses of ethyl 2-(4-(4-(4-trifluoromethyl)benzamido)-1,2,5-oxadiazol-3-yl)phenoxyacetate (24) and 2-(4-(4-(4-trifluoromethyl) benzamido)-1,2,5-oxadiazol-3-yl)phenoxyacetic acid (25). To a solution of compound 21 (0.088 mmol) in dry N-N-dimethylformamide (0.7 ml), K2CO3 (0.177 mmol) and ethyl chloroacetate (0.088 mmol) were added. The reaction mixture was stirred under nitrogen atmosphere at room temperature for 24 h, and then filtered. The filtrate was concentrated under reduced pressure and dissolved in ethyl acetate (2 ml). The organic layer was washed with 1N NaOH and brine, dried over Na2SO4, filtered and evaporated in vacuo, obtaining compound 24. Yield: 89%. Off-white foam. 1H-NMR (acetone-d6): δ 1.26 (t, J=7.1 Hz, 3H, CH3), 2.81 (br s, 1H, NH exchanged with D2O), 4.23 (q, J=7.1 Hz, 2H, CH2), 4.81 (s, 2H, CH2), 7.09 (d, J=9.0 Hz, 2H, ArH), 7.71 (d, J=8.5 Hz, 2H, ArH), 8.38-8.49 (m, 4H, ArH) ppm. 13C-NMR (acetone-d6): δ 14.04, 61.60, 65.55, 115.29, 118.39-121.97-125.54-129.11 (q), 122.34, 126.28, 128.63, 128.89, 133.30-133.73-134.16-134.59 (q), 134.05, 147.24, 153.84, 159.16, 166.50, 168.53 ppm. 19F-NMR (CDCl3): δ -63.59 (s, CF3) ppm. MS (ESI): calcd. for C20H16F3N3O5 435, found 436 [M+H]+.
Compound 24 was dissolved in tetrahydrofuran/ethanol (1:1.5 ml) and 1N NaOH was added dropwise. The reaction was refluxed for 30 min. and the solvents were evaporated under reduced pressure. The residue was diluted with water and washed with dichloromethane. The aqueous layer was acidified with 1N HCl and extracted with ethyl acetate. The organic phase was dried over Na2SO4, filtered and concentrated under vacuum, affording compound 25 (13). Yield: 93%. Light yellow foam. 1H-NMR (acetone-d6): δ 2.81 (br s, 1H, NH exchanged with D2O), 4.80 (s, 2H, CH2), 7.08 (d, J=9.0 Hz, 2H, ArH), 7.81 (d, J=9.0 Hz, 2H, ArH), 7.93 (d, J=8.1 Hz, 2H, ArH), 8.26 (d, J=8.1 Hz, 2H, ArH), 10.44 (br s, 1H, OH exchanged with D2O) ppm. 13C-NMR (acetone-d6): δ 65.35, 115.26, 118.40-121.96-125.54-129.12 (q), 122.31, 126.25, 128.65, 128.95, 133.61-134.03-134.45-134.88 (q), 134.19, 147.25, 153.75, 159.23, 166.56, 170.15 ppm. 19F-NMR (CDCl3): δ-63.61 (s, CF3) ppm. MS (ESI): calcd. for C18H12F3N3O5 407, found 406 [M-H]−.
Synthesis of dibenzyl(4-(4-(4-trifluoromethyl)benzamido)-1,2,5-oxadiazol-3-yl)phenylphosphate (28). Compound 21 (0.143 mmol) was dissolved in dry acetonitrile (1 ml) under nitrogen atmosphere and the solution was cooled to −10°C. Subsequently, CCl4 (0.07 ml), N,N-diisopropylethylamine (0.301 mmol) and N,N-dimethylaminopyridine (0.014 mmol) were added. Then, dibenzylphosphite (0.208 mmol) was dripped, and the mixture was stirred at −10°C for 3 h. A solution of 0.5M K2HPO4 (1 ml) was added, and the reaction was allowed to warm at room temperature. The mixture was extracted with ethyl acetate (3×1 ml) and the combined organic phases were washed with water and brine, dried over Na2SO4, filtered and concentrated under reduced pressure. Compound 28 was purified by flash chromatography (eluent mixture: cyclohexane/ethyl acetate, 90:10). Yield: 49%. Off-white foam. 1H-NMR (acetone-d6): δ 5.17 (s, 2H, CH3), 5.22 (s, 2H, CH2), 7.30-7.45 (m, 9H, ArH), 7.85 (d, J=8.7 Hz, 2H, ArH), 7.93 (d, J=8.1 Hz, 2H, ArH), 8.27 (d, J=8.1 Hz, 2H, ArH), 10.59 (br s, 1H, NH exchanged with D2O) ppm. 13C-NMR 69.50, 118.39, 119.60, 120.27, 121.93, 125.54, 126.26, 128.30, 128.56, 129.11, 132.21, 133.90-134.34-134.76-135.20 (q), 138.59, 147.39, 149.53, 151.81, 165.46 ppm. 19F-NMR (CDCl3): δ -63.80 (s, CF3) ppm. MS (ESI): calcd. for C30H23F3N3O6P 609, found 610 [M+H]+.
Antiproliferative assay on HCT-116 cell line
Cell line culture. HCT-116 (human colorectal cancer) cell line was obtained from Cell-Lab (Unimore) and cultured in Dulbecco's modified Eagle medium (DMEM) medium (Euroclone, Devon, UK), supplemented with 10% heat-inactivated fetal bovine serum (Euroclone) and 1% Pen/Strep (Euroclone). Cultures were equilibrated with humidified 5% CO2 in air at 37°C. All studies were performed in Mycoplasma-negative cells, as routinely determined with the MycoAlert Mycoplasma detection kit (Lonza, Walkersville, MD, USA).
Cytotoxicity screening. Cytotoxicities of the compounds used in the present study were determined by the MTT assay (14). Briefly, the cells were seeded into 96-well plates and cultured overnight. Various concentrations of the test compounds, dissolved in DMSO, were then added and incubated for 48 h. After incubation, 10% V/V of a solution of 0.5 mg/ml MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide; Sigma-Aldrich) was added and incubation at 37°C continued for 3 h. The medium was removed and 100 μl DMSO was added to dissolve the dark blue crystals. After incubation for 30 min at room temperature to ensure that all crystals were dissolved, absorbance was measured using an ELISA plate reader (TECAN, Grödig, Salzburg, Austria) at 595 nm.
Antiproliferative assay on HeLa cell line
Cell line culture. HeLa (human cervix adenocarcinoma) cells were grown in Nutrient Mixture F-12 [HAM] (Sigma Chemical Co.). 10% Heat-inactivated fetal calf serum (Invitrogen), 100 U/ml penicillin, 100 μg/ml streptomycin and 0.25 μg/ml amphotericin B (Sigma Chemical Co.) were added to the medium. The cells were cultured at 37°C in a moist atmosphere of 5% CO2 in air.
Cytotoxicity screening. HeLa cells (4-5×104) were seeded into a 24-well cell culture plate. After incubation for 24 h, various concentrations of the test agents were added, and the cells were then incubated in standard conditions for a further 48 h. A trypan blue assay was performed to determine cell viability. Cytotoxicity data were expressed as IC50 values, i.e. the concentration of the test agent inducing 50% reduction in cell number compared with control cultures.
Topoisomerases-mediated DNA relaxation assays. Supercoiled pBR322 plasmid DNA (0.25 μg, Fermentas Life Sciences) was incubated with 1U topoisomerase II (human recombinant topoisomerase II α, USB Corporation) or 2U topoisomerase I (human recombinant topoisomerase I, TopoGen) and the test compounds as indicated, for 60 min at 37°C in 20 μl reaction buffer. Reactions were stopped by adding 4 μl stop buffer (5% sodium dodecyl sulfate (SDS), 0.125% bromophenol blue, and 25% glycerol), 50 μg/ml proteinase K (Sigma-Aldrich) and incubating for a further 30 min at 37°C. The samples were separated by electrophoresis on a 1% agarose gel at room temperature. The gels were stained with 1 μg/ml ethidium bromide in TAE buffer (0.04 M Tris-acetate and 0.001 M EDTA), transilluminated by UV light, and fluorescence emission was visualized by a CCD camera coupled to a Bio-Rad Gel Doc XR apparatus.
Docking studies. Gold software (https://www.ccdc.cam.ac.uk/solutions/csd-discovery/components/gold/; version 5.6) was used to carry out the docking of the oxadiazole derivatives into the X-ray structures of topoisomerase I (PDB code: 1K4T) using default parameter setting. All docking calculations were performed on an Intel I Core I i7-2600 CPU, using Windows 7 operating system. The putative binding mode of the ligands was predicted by genetic algorithm (GA) search approach and ChemPLP scoring function. The binding site was defined from the co-crystalized ligand considering a radius of 6 Å during docking calculations. Ligand structures were sketched and energy-minimized by Sybyl 8.1 (Tripos, Inc., St. Louis, MO, USA) on an Intel I Core I2 Duo CPU E8500, using Ubuntu 14.04 operating system. For each ligand, ten poses were generated and energetically ranked. All complexes obtained were analyzed to identify the most likely solution in terms of ligand-protein interactions.
Results
Chemistry. In this section, the synthetic procedures (Figure 2) to obtain the new products (14-17, 24-26 and 28) are reported; the remaining compounds were obtained through previously reported methods (3). Briefly, the key intermediates 30a-c, synthesized from the properly substituted chlorobenzaldehydes following a multi-step procedure (4), led to the final products 15, 16 and 26, through a coupling reaction with the suitable acyl chlorides. The same procedure was applied to obtain other intermediates: in detail, intermediate 31 was hydrolyzed to afford 13, which was subsequently esterified to give 14. Compound 21, achieved by debenzylation of 27 (3), was protected to give 28 and 24. The latter was hydrolyzed to afford 25. Finally, 17 was obtained by reductive amination of the key intermediate 30a.
Antiproliferative activity on HCT-116 and HeLa cell lines. To complete the available data concerning the activity of our library members, the cytotoxicity of the new compounds (14-17, 24-26 and 28) was tested on HCT-116 cell line by the MTT assay (Table I and Figure 3).
Furthermore, with the aim of widening the antiproliferative profile, the entire library of 1,2,5-oxadiazole derivatives was also tested on HeLa, a human tumor cell line. MD77 was taken as reference compound (Table I). The results highlighted that among the new compounds, 16, 25 and 28 exhibited a detectable antiproliferative effect on HCT-116 cell line, comparable to that of some previously synthesised derivatives, but lower with respect to that of the reference compound MD77.
Concerning the antiproliferative activity on HeLa cell line, the obtained IC50 values were in agreement with those obtained on HCT 116. Interestingly, some derivatives were able to exert a significant antiproliferative effect, with IC50 values below 20 μM (6, 19 and 29), on both HCT-116 and HeLa cell lines. These results suggest a crucial role for the concurrent insertion of CF3 and Cl groups in position meta and para of the phenyl rings. Moreover, the IC50 values reported in Table I highlight a detrimental role in the biological activity for the ortho position, as a notable increase was observed for derivatives 5 and 18. Similarly, the antiproliferative capacity appears negatively affected by the absence of the CF3 group (comparing 4 vs. 19). Finally, the contribution of the CF3 to cytotoxicity seems more important with respect to that of Cl, since derivative 20 exerted a higher antiproliferative effect with respect to 7 on both cell lines.
AlphaScreen-based assay. Considering that several examples of oxadiazole derivatives targeting the SH2 domain of STAT3 have been reported (15, 16), the compounds endowed with the most interesting antiproliferative activity were tested by the AlphaScreen-based assay to evaluate their potential inhibition of the interaction between the SH2 domain and pTyr-containing peptides, at 30 μM concentration. The obtained data indicated that the derivatives did not interact with the SH2 domain (unpublished data).
Effect of oxadiazole derivatives of topoisomerase-mediated DNA relaxation. In the attempt to identify the molecular target responsible for the antiproliferative effect of the compounds and taking into account that some oxadiazole derivatives were shown to inhibit topoisomerases (10, 11), compounds 1-29 and MD77 were tested versus the supercoiled DNA relaxation activity of topos I and II.
As for topo I, all the derivatives that showed an inhibitory effect on the catalytic activity are shown in Table II. Interestingly, the most active derivative within the subseries (3) is characterized by the presence of only a Cl atom in ortho position. The shift from ortho (3) to meta (4) provokes a decrease in the activity, while the inhibitory effect is completely abolished when the Cl is inserted in the para position (compound 2, not shown in Table II due to its inactivity in the assay). Nevertheless, the detrimental effect of the p-Cl can be partially overcome when a carboxylic acid is present in p-R1 (13). A dihydrogen phosphate in the same position (12) induces only a weak inhibition, much like the CF3 group in ortho (5) and meta (6). Furthemore, the unfavorable influence of the CF3 is supported by the inefficacy of the reference compound, MD77.
IC50 values (μM) of the compounds against the human colorectal cancer HCT-116 and human cervix adenocarcinoma HeLa cell lines. The IC50 is defined as the concentration causing 50% growth inhibition in treated cells when compared to control cells after 48 h drug exposure. Values are means±SD of three separate experiments performed in quadruplicate.

Inhibition of cell growth. HCT-116 cells were allowed to attach overnight and then treated with the indicated concentrations of compounds 14-17, 24-26 and 28 for 48 h. Cell survival percentages are the mean±SD of three separate experiments performed in quadruplicate.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
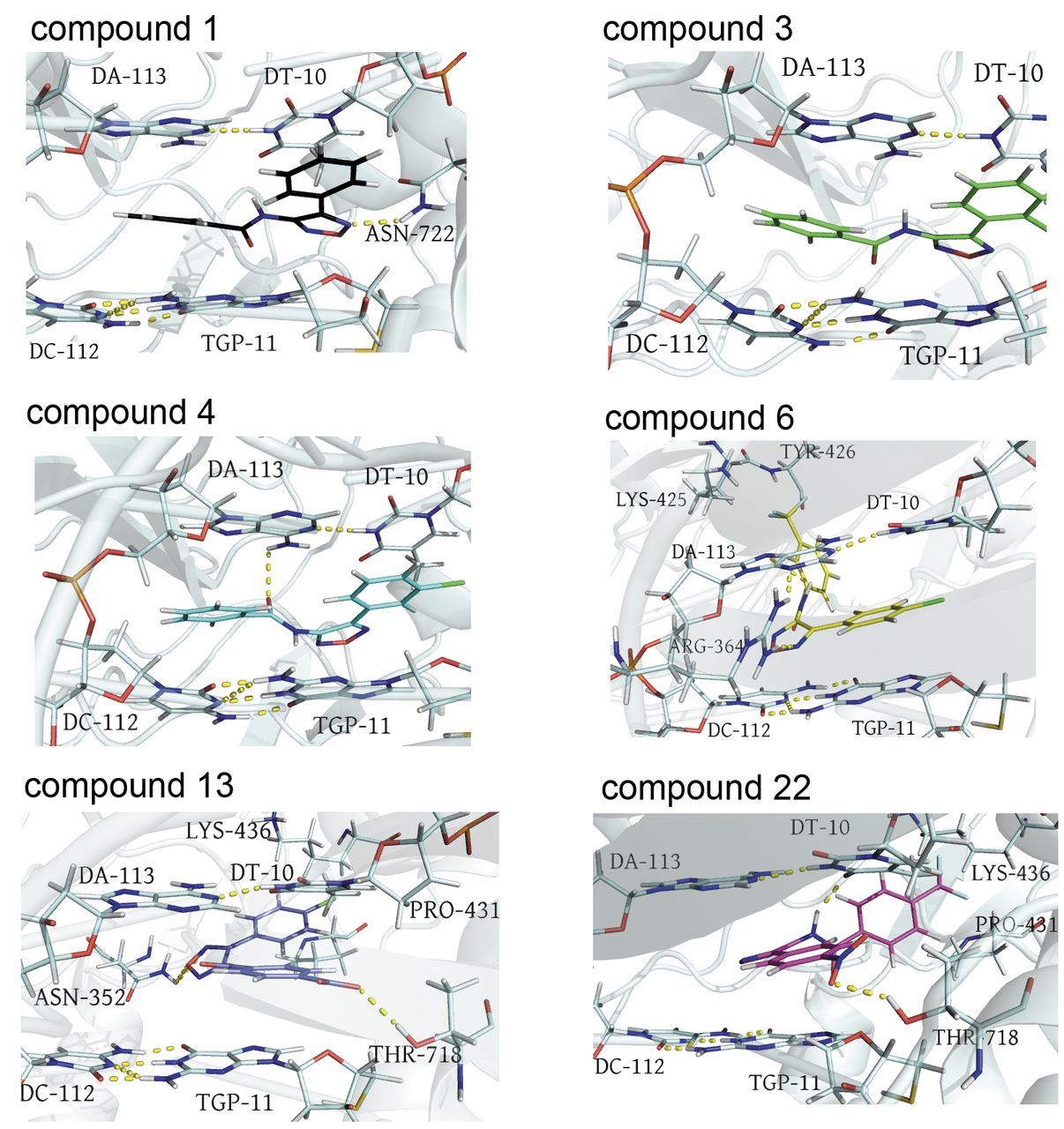
Docking poses for compound 1, 3, 4, 6, 13 and 22 into topo I (1K4T) interaction site. Ligands, DNA bases and key amino acids for ligand interactions are in stick. H-bonds are shown as yellow dotted lines.
Effect of the tested compounds on the relaxation of supercoiled plasmid DNA by topoisomerase I.
Considering the subseries having the p-CF3 in R1, only compound 22 is able to exert a significant inhibitory activity suggesting a relevant role for electron-withdrawing substituents.
Finally, it should be noted, that the unsubstituted 1 shows a significant inhibitory effect on topo I activity, thus confirming the role of the oxadiazole ring as a potential scaffold for the development of anti-topo I agents.
A comparison between the antiproliferative data (Table I) and the ability to inhibit topo I relaxation activity (Table II) highlights for 1-29 the lack of a clear relationship among the two effects. Nevertheless, these results suggest that the 1,2,5 oxadiazole ring could represent a suitable scaffold for the development of topo I inhibitors, taking into account that the cellular uptake and/or metabolic pathways could play an important role for the delivery to the possible intranuclear target.
Finally, compounds 1-29 are not able to significantly inhibit the relaxation of supercoiled DNA mediated by topo II at the same concentration (data not published).
Docking studies. To clarify the mode of action of this class of compounds against topo I, docking studies were performed considering active (1, 3, 4, 13 and 22), inactive (MD77, 29) and weakly active (6) derivatives. PDB (Protein Data Bank) structure 1K4T (topo I co-crystallized with topotecan) was chosen to model the binding of the compounds to the target. The binding sites were defined at the level of DNA cleavage. The docking poses of compounds 3, 4, 6 and 22, exhibiting interesting interactions with topo I, are reported in Figure 4. Docking scores of the compounds tested as topo I inhibitors are reported in Table III.
Docking scores of compounds 1, 3, 4, 6, 13, 22, 29 and MD77 for interaction with topo I.
The highest docking scores were observed for compounds 3, 4, 6 and 22, where π-π stacking interactions, H-bonds and π-alkyl interactions (17) contribute to stabilize the complexes between the target macromolecule and the ligand. The top-scored pose of 3 shows a good orientation at the binding site, indicating the ability to intercalate between the upstream (−1) and downstream (+1) base pairs to form base-stacking interactions with both the −1 (upstream) and +1 (downstream) base pairs. In detail, the phenyl-oxadiazole core of 3 interacts by π-π stacking interactions with DT10 and TGP11 (TGP11=5’-Thio-2’-deoxy-guanosine phosphonic acid). Moreover, compound 3 is stabilized though the phenyl substituent (R1) by π-π stacking interactions with DA113, DC112, and TCP11; moreover, one H-bond (N-H--π) (18) with DA113 can be observed. Additionally, the Cl atom (R) of compound 3 binds TGP11 and DT10 by π-alkyl interactions (17).
Changing the Cl atom from the ortho to the meta position, as in compound 4, weakly affects the binding affinity, as revealed by the in vitro results. In this molecule (4), the same pattern of interactions was found, with the only exception of that with TGP11.
As expected, the introduction of a –NO2 group (R) and a CF3-Ph (R1) moiety at the para-position of the phenyl-oxadiazole scaffold as in compound 22, determines a different orientation of the ligand into the binding site, resulting in a partial intercalation of 22 between the double stranded DNA. A π-π T shaped interaction with DT10 and compound 22 also occurs. The CF3-Ph moiety in 22 contributes to form a protein-ligand complex by π-alkyl (17) and alkyl-alkyl interactions with Pro431 and Lys436. In addition, it is hydrogen bonded to Lys436 (CH--F) (19). Furthermore, the p-NO2 substituent of 22 is involved in H-bonding interactions with Thr718 (OH…ON) and DT10 (CH–O) (20). Additionally, the phenyl-oxadiazole core of 22 is anchored to the target protein by π-π stacked interactions with DT10, DA113 and TGP11. However, one unfavourable interaction (CONH---HN unfavourable donor-donor) with DA113 also occurs in 22.
The loss of π-π interactions of the phenyl substituent (R1) in 6, due to the shift of the CF3-Ph (R1) moiety from the para position to meta position, might justify the reduction in the inhibitory activity.
However, the Cl atom of 6 shares π-alkyl interactions with DT10 and TGP11. The CF3 group in R1 is involved in a halogen H-bond with Tyr426 and Lys 425 (21), and a hydrophobic interaction occurs between the carbon atom of -CF3 and Lys425. The phenyl-oxadiazole skeleton is able to give π-π stacked interactions with DC112, TGP11, DA113, DT10 and a H-bond with Arg364.
A lower binding affinity, as estimated by lower docking scores, was found for compounds 1, 13, 29 and MD77. With the only exception of ligand 1, a minor involvement in π-π interactions was observed. Moreover, the introduction of a Cl atom or -CF3 group at the para or meta position (R) of the phenyl-oxadiazole nucleus as in MD77 and 16, along with the presence of a -CF3Ph (MD77) or a COOH-Ph (13) group, in the para position of benzamide, seems to be responsible for a decrease in the number of hydrogen bonds and π-π staking interactions. Through its phenyl substituent in R1, ligand 1 forms π-π stacking interactions with DA113 and TGP11. In addition, considering the phenyl-oxadiazole core of 1, it π-πstacks TGP11 and DT10, and a H-bond occurs between the oxadiazole nitrogen and the amine group of Asn722.
The carboxyl group (R1) of 13 is stacked to DT10, TGP11 and it is H-bonded to Thr718 (CO--OH; O—HC) (20). In addition to a H-bond between the phenyl-oxadiazole skeleton of 13 and Asn352 (N--HN), hydrophobic interactions also occur between the Cl atom (R) and Lys436 and Pro431. Furthermore, 29 does not intercalate between DNA base pairs, but binds to the protein in a different site, compared to topotecan. For this reason, the in vitro inhibitory activity of 29 might be due to different modes of action.
Discussion
During our ongoing research we aimed at the identification of novel antiproliferative compounds, possibly endowed with high activity towards different cancer cell lines, several 1,2,5-oxadiazole derivatives inhibiting HCT-116 cell line growth have recently been discovered. To complete our small library, some novel compounds were designed, synthesized and tested (14-17, 24-26 and 28 along with the previously synthesized 27, 29 and MD77). Then, to further strengthen our findings, the antiproliferative effects of all oxadiazole derivatives on HeLa cell line were evaluated and the data confirmed the previous interesting trends. In particular, compounds 6, 19 and 29 gain a significant antiproliferative activity, with IC50 values below 20 μM, on both HCT-116 and HeLa cell lines (Table I). Moreover, cytotoxicity values show a detrimental role in the biological activity for the ortho position, as for 5 and 18 derivatives against HCT-116 and HeLa cell lines (Table I) and the presence of the CF3 seems to have a more remarkable effect with respect to that of Cl, as an important increase is shown by derivative 20 with respect to 7 on both cell lines. With the aim of identifying their possible target, a careful investigation on the role of the oxadiazole scaffold in the field of antitumor chemotherapy was performed; in light of our findings, we decided to assay the inhibitory activity of all compounds on the SH2 domain of STAT3 and on topos I and II. None of the compounds was active on the SH2 domain of STAT3 and topo II, at the tested concentration. Instead the preliminary results highlighted that several derivatives (in particular 3, 22 and, to a lesser extent, 1, 4 and 13) inhibited the catalytic activity of topo I. The latter is involved in the control of DNA stability during DNA replication, transcription and recombination and is recognized as the target of anticancer drugs, that act by stabilizing the enzyme–DNA cleavable complex leading to cell death (22). Docking studies supported these results, although, unexpectedly, some compounds (1 and 13) did not exhibit a relevant antiproliferative activity towards the tested cancer cell lines. In particular, no clear relationship could be established between the antiproliferative and anti-topo I effect, suggesting that they could interact with other targets. Further studies will be carried out to assess the primary mechanism of action of these compounds.
Conclusion
Considering that some of the most important topo I inhibitors in therapy are outdated and endowed with pharmacokinetic problems and adverse effects (23, 24) and few new inhibitors have been reported in the literature, the inhibitory effect exerted by some of the 1,2,5-oxadiazoles towards topo I relaxation activity makes this class of derivatives remarkably interesting. In fact, topo I inhibitors were introduced in recent years into the clinical setting as promising antineoplastic agents (25). In particular, our results could represent a promising strategy to develop properly functionalized new 1,2,5-oxadiazole derivatives able to target topo I and to overcome kinetics and/or cell metabolic drawbacks.
Acknowledgements
This research was financially supported by University of Milan and the Italian MIUR (PRIN Research Project grant no. 20105YY2H-007, Prof. D. Barlocco).
- Received November 8, 2018.
- Revision received November 20, 2018.
- Accepted November 21, 2018.
- Copyright© 2019, International Institute of Anticancer Research (Dr. George J. Delinasios), All rights reserved