

Abstract
Background/Aim: The aim of this study was to evaluate the effect of evodiamine alone or in combination with chemotherapeutic agents on thyroid carcinoma cells. Materials and Methods: TPC-1 and SW1736 thyroid carcinoma cells were used. Cell viability, cytotoxic activity, apoptosis and migration were examined by applying appropriate methods. Drug combination analysis was performed. Results: Evodiamine treatment of cells decreased cell viability, and Bcl2 and phospho-AKT protein levels. Cytotoxic activity and the percentage of apoptotic cells increased. After co-treatment of wortmannin, cell viability, and phospho-AKT and Bcl2 protein levels decreased, and cytotoxic activity increased. In transforming growth factor-β-treated cells, evodiamine attenuated variations in morphology, growth and migration, and increased p21 and p53 protein levels, and decreased β-catenin, N-cadherin, vimentin, phospho-AKT, matrix metalloproteinase-2 and matrix metalloproteinase-9 protein levels. When cells were treated with both evodiamine and chemotherapeutic agents, all combination index values were lower than 1.0. Conclusion: Evodiamine was cytotoxic towards thyroid carcinoma cells, and repression of AKT reinforced evodiamine-induced cytotoxicity. Furthermore, evodiamine ameliorated proliferation, migration and epithelial–mesenchymal transition, and synergized with chemotherapeutic agents.
- Thyroid carcinoma
- evodiamine
- AKT
- chemotherapeutic agent
- synergism
Well differentiated thyroid cancer (WDTC), including papillary thyroid cancer (PTC) and follicular thyroid cancer (FTC), is the most frequent subtype with excellent outcomes, whereas it is refractory to radioactive iodine (RAI) therapy in approximately two thirds of patients with distant metastasis (1-3). On the other hand, undifferentiated thyroid cancer (UDTC) including anaplastic thyroid cancer (ATC) presents highly aggressive features characterized as extrathyroidal invasion and distant metastasis with disastrous outcomes (1-3). Since patients with RAI therapy-refractory WDTC and UDTC are not responsive to conventional treatment modalities, new therapeutic approaches to improve therapeutic efficacy against cancer cells are under investigation (1-3).
Evodiamine is a natural indole alkaloid isolated from the fruit of Evodia rutaecarpa, also named as Wu-Zhu-Yu, and a multi-target compound possessing a broad spectrum of biological actions (4). Evodiamine has been traditionally used for treatment of abdominal pain, vomiting, diarrhea, headache and postpartum hemorrhage in herbal medicine (4). Evodiamine exerts beneficial activities in thermoregulation, nociception, inflammation, obesity, cardiovascular disease, infectious disease and Alzheimer's disease, and poses antitumor properties in a variety of cancer cells (4, 5). It has been proposed that the mechanism of anticancer action of evodiamine is cell cycle arrest at G2/M phase through activation of Cdc2/cyclin B complex, and induction of cell death by modulating survival-related proteins including Bcl2 family proteins, p21 and p53 as well as multiple signaling pathways including PI3K/AKT and nuclear factor-kappaB (NF-ĸB) (6-15). However, the influence of evodiamine on survival of thyroid carcinoma cells has not been identified.
Evodiamine attenuates migration, invasion and metastasis of cancer cells, and thereby abrogates cancer progression (5, 16). Evodiamine represses cell migration and invasion via regulation of matrix metalloproteinase-2 (MMP-2) and matrix metalloproteinase-9 (MMP-9) in prostate and nasopharyngeal cancer cells, and reduces formation of metastatic foci in colon cancer animal model (17-19). In regard to epithelial–mesenchymal transition (EMT), evodiamine suppresses hepatocyte growth factor (HGF)-activated invasiveness of cancer cells, and abolishes self-renewal through mitigation of EMT in gastric cancer stem cells, and ameliorates transforming growth factor-β(TGF-β)-activated EMT in in vitro and in vivo models (15, 20-22). Meanwhile, evodiamine has cytotoxic activities in breast cancer refractory to conventional chemotherapeutic agents (23, 24). Furthermore, evodiamine exhibits cytostatic and cytotoxic properties in paclitaxel-resistant ovarian cancer cells (25). However, the combined effect of evodiamine with chemotherapeutic agents on thyroid carcinoma cells has not been explored.
The aim of the present study was to assess the influence of evodiamine on cell survival, proliferation, migration and EMT, and analyze the effect of evodiamine in combination with chemotherapeutic agents on growth and survival in thyroid carcinoma cells. Our results demonstrated that evodiamine has a negative impact on cell survival, proliferation, migration and EMT, and has synergistic action with chemotherapeutic agents in inhibition of growth and survival of thyroid carcinoma cells.
Materials and Methods
Materials. RPMI1640, fetal bovine serum (FBS), L-glutamine and streptomycin/penicillin were purchased from Life Technologies (Carlsbad, CA, USA). Evodiamine, doxorubicin, paclitaxel and cisplatin were obtained from BioVision (Linda, CA, USA), and dissolved in dimethylsulfoxide (DMSO). Control cells were treated with 0.1% vehicle DMSO. The primary antibodies against Bcl2, Bax, survivin, phospho-histone H2A.X (γH2AX), cleaved poly (ADP-ribose) polymerase (PARP), total and phospho-NF-ĸB (Ser536), total and phospho-c-Jun N-terminal kinase (JNK) (Thr183/Tyr185), total and phospho-extracellular signal-regulated kinase (ERK) 1/2 (Thr402/Tyr404), p21, p53, MMP-2, MMP-9, β-catenin, N-cadherin, vimentin and slug were purchased from Cell Signaling Biotechnology (Danvers, MA, USA). The primary antibodies against total and phospho-AKT (Ser473) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA), and the primary antibody against β-actin was from Sigma (St. Louis, MO, USA). All other reagents were purchased from Sigma unless otherwise stated.
Cell culture. For experiments, TPC-1 human PTC cells were obtained from Professor Young Joo Park (Division of Endocrinology and Metabolism, Seoul National University, Republic of Korea), and grown in RPMI1640 supplemented with 10% heat-inactivated FBS and 1% streptomycin/penicillin. SW1736 human ATC cells were purchased from Cell Lines Service (CLS GmbH, Eppelheim, Germany), and grown in RPMI1640 supplemented with 2 mM L-glutamine, 10% heat-inactivated FBS and 1% streptomycin/penicillin. Cells received fresh medium at regular intervals. Treatments and experiments were performed using cells that were 70% confluent.
CCK-8 assay. Cell viability was determined by the CCK-8 Assay Kit (Dojindo laboratories, Kumamoto, Japan). Cells (5×103/100 μl) in each well on 96-well plates were incubated overnight, and treated with agents for an additional 4 h at 37°C. Absorbance was measured using Glomax™ Discover System GM3000 (Promega, Madison, WI, USA). All experiments were performed in triplicate.
Cytotoxicity assay. Cytotoxic activity was measured by the LDH Cytotoxicity Assay Kit (BioVision, Linda, CA, USA). Cells (5×103/100 μl) in each well on 96-well plates were incubated, and centrifuged at 250 g for 10 min. Supernatant of 100 μl was transferred in clear 96-well plates. After addition of reaction mixture (2.5 μl Catalyst solution in 112.5 μl Dye solution), cells were incubated for 30 min at room temperature. Absorbance was measured using Glomax™ Discover System GM3000 (Promega). All experiments were performed in triplicate.
FACS analysis. Apoptotic cells were analyzed by the Annexin V-FITC Apoptosis Detection Kit (BD Biosciences Pharminogen, San Diego, CA, USA). Cells (1×105/ml) in each well on 6-well plates were incubated, harvested, washed, and fixed according to manufacturer's protocol. FITC annexin V and/or propidium iodide (PI) in 1× binding buffer was added for 15 min at room temperature, and analysis was made using a CytoFLEX™ Flow Cytometer (Beckman Coulter Inc., Brea, CA, USA) and CytExpert Software (Beckman Coulter Inc., Brea, CA, USA). All experiments were performed in triplicate.
Cell number count. Cell growth was determined by cell number count using trypan blue. Cells (1×105/500 μl) in each well on 12-well plates were incubated, and mixed with trypan blue dye at 37°C. Stained cells were counted using a hemocytometer. All experiments were performed in triplicate.
Wound healing assay. Cell migration was measured by the CytoSelect™ 24-Well Wound Healing Assay Kit (Cell Biolabs, San Diego, CA, USA). Following generation of a wound field (0.9 mm), cells were incubated, and treated at 37°C. The wound closure was monitored by light microscope, and the cell migration rate was calculated according to the following equation: cell migration= [length of cell migration (nm)/migration time (h)]. All experiments were performed in triplicate.
Western blotting. The total protein was extracted by RIPA buffer (Sigma) containing 1× protease inhibitor cocktail and 1× phosphatase inhibitor cocktail set V (Calbiochem, La Jolla, CA, USA). Western blotting was performed using specific primary antibodies and horseradish peroxidase-conjugated anti-rabbit and anti-mouse secondary antibodies. Bands were detected using ECL Plus Western Blotting Detection System (Thermo Fisher Scientific, Rockford, IL, USA). The protein levels were quantified by densitometry using ImageJ software (NIH), and normalized to β-actin levels. The relative levels of protein to β-actin were obtained. All experiments were performed in triplicate.
Drug combination analysis. Combination index (CI) and isobologram were calculated by CalcuSyn program version 2.11 (Biosoft, Great Shelford, Cambridge, UK), and the effect of drug interactions was quantitatively documented. CI values less than 1.0, 1.0 and greater than 1.0 manifest synergism, additivity and antagonism, respectively. The isobologram is formed by plotting the doses of each agent required for 50% inhibition (ED50) on the x- and y-axis, and connecting them to draw a line segment, which is ED50 isobologram. Combination data points that fall on, below and above the line segment manifest additivity, synergism and antagonism, respectively. All combinations were performed in triplicate.
Statistical analysis. All data are expressed as mean±standard error (S.E). Data were analyzed by unpaired Student's t-test or ANOVA as appropriate. A p-value less than 0.05 was considered to indicate statistical significance. All analyses were performed using SPSS program version 24.0 (SPSS, Chicago, IL, USA).

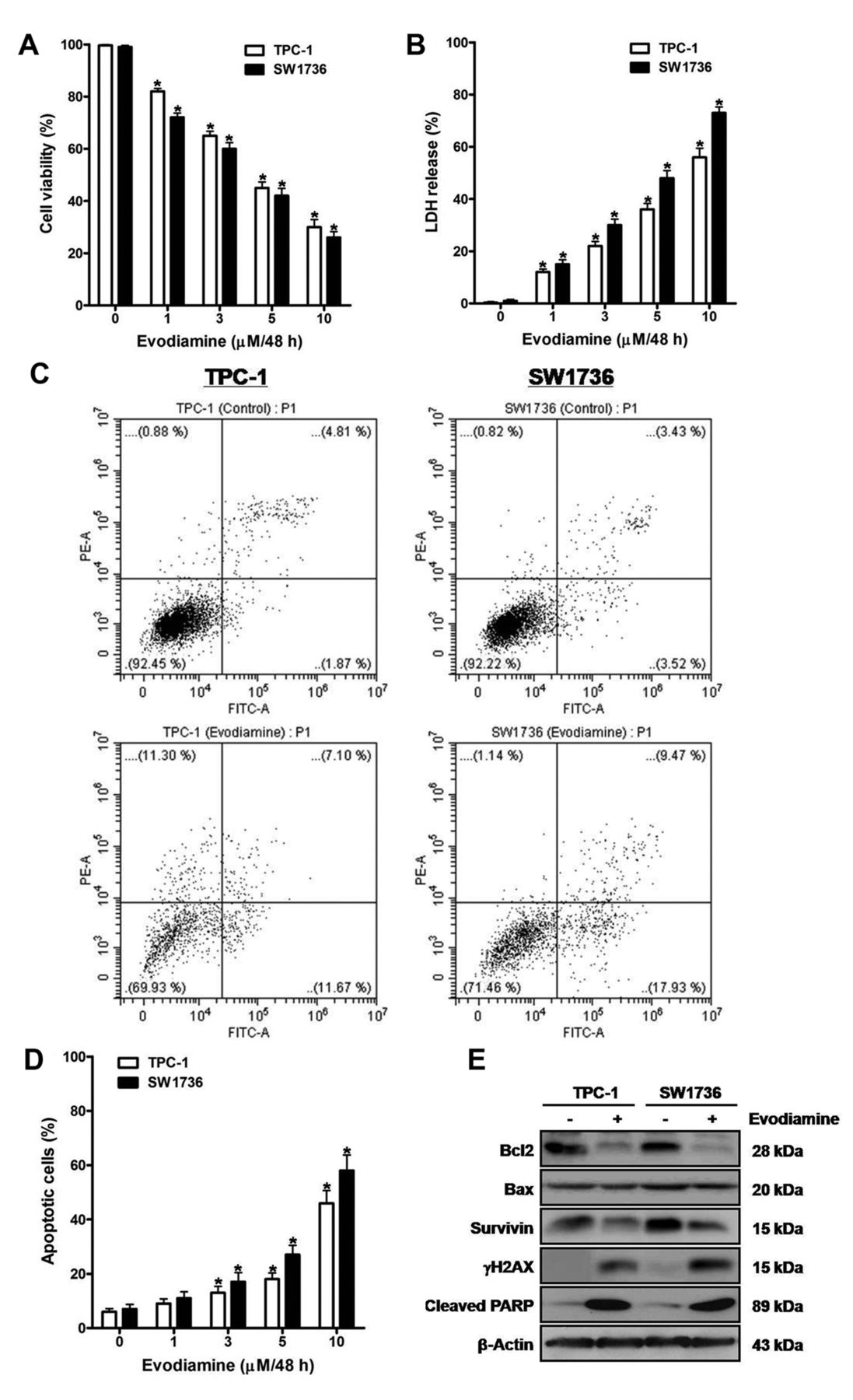
The effect of evodiamine on survival of thyroid carcinoma cells. TPC-1 and SW1736 cells were treated with 1, 3, 5 and 10 μM evodiamine for 48 h, and cell viability (A) and cytotoxic activity (B) were measured using CCK-8 assay and cytotoxicity assay, respectively. C: TPC-1 and SW1736 cells were treated with 5 μM evodiamine for 48 h, and then the percentage of apoptotic cells was measured using FACS analysis. D: TPC-1 and SW1736 cells were treated with 1, 3, 5 and 10 μM evodiamine for 48 h, after which the percentage of apoptotic cells was measured. E: TPC-1 and SW1736 cells were treated with 5 μM evodiamine for 48 h, and the protein levels of Bcl2, Bax, survivin, γH2AX and cleaved PARP were measured. All experiments were performed in triplicate. The blots are representative of three independent experiments. Data are expressed as mean±S.E. *p<0.05 vs. each matched control.
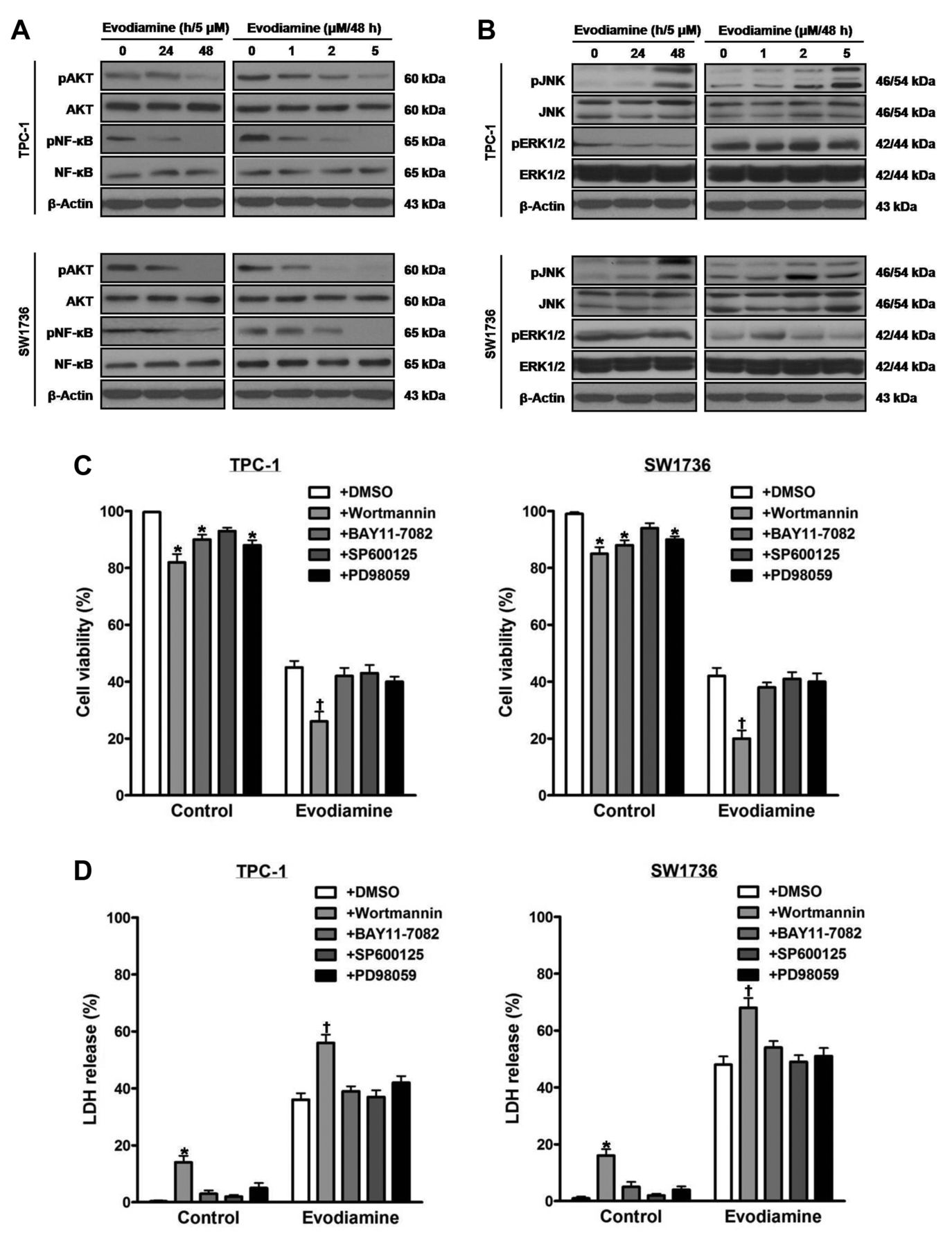

The influence of evodiamine on signal proteins in thyroid carcinoma cells. TPC-1 and SW1736 cells were treated with 5 μM evodiamine for 24 and 48 h, and 1, 2, and 5 μM for 48 h, and the total and phospho-protein levels of AKT and NF-kB (A) as well as those of JNK and ERK1/2 (B) were measured. TPC-1 and SW1736 cells were treated with 2 μM of the PI3K inhibitor wortmannin, 0.2 μM of the NF-κB inhibitor BAY11-7082, 2 μM of the JNK inhibitor SP600125 or 2 μM of the MEK inhibitor PD98059 prior to treatment with 5 μM evodiamine for 48 h, and then cell viability (C) and cytotoxic activity (D) were measured. E and F: TPC-1 and SW1736 cells were treated with 2 μM wortmannin before treatment with 5 μM evodiamine for 48 h, after which the protein levels of total and phospho-AKT, Bcl2, Bax and cleaved PARP were measured, and quantified by densitometry, and normalized to β-actin levels. All experiments were performed in triplicate. The blots are representative of three independent experiments. Data are expressed as mean±S.E. *p<0.05 vs. each matched control; †p<0.05 vs. cells treated with both evodiamine and DMSO; ‡p<0.05 vs. cells treated with evodiamine alone.
Results
Evodiamine induces death of thyroid carcinoma cells. To evaluate the effect of evodiamine on cell survival, TPC-1 and SW1736 cells were treated with 1, 3, 5 and 10 μM evodiamine for 48 h, and then cell viability (Figure 1A) and cytotoxic activity (Figure 1B) were measured using the CCK-8 and cytotoxicity assays, respectively. Following treatment, cell viability was diminished, and cytotoxic activity was enhanced in a dose-dependent manner.
When cells were treated with 5 μM evodiamine for 48 h, the percentage of apoptotic cells using FACS analysis was enhanced (Figure 1C). Furthermore, treatment of cells with 1, 3, 5 and 10 μM evodiamine for 48 h enhanced the percentage of apoptotic cells in a dose-dependent manner (Figure 1D).
To estimate the influence of evodiamine on the expression of survival-related proteins, extracts of cells treated with 5 μM evodiamine for 48 h were examined by western blot using antibodies against Bcl2, Bax, survivin, γH2AX and cleaved PARP (Figure 1E). After treatment, the protein levels of γH2AX and cleaved PARP were enhanced, and those of Bcl2 and survivin were diminished without alteration in Bax protein levels.
Inactivation of AKT intensifies evodiamine-induced death of thyroid carcinoma cells. In order to determine the impact of evodiamine on signal proteins, extracts of cells treated with 5 μM evodiamine for 24 and 48 h, and 1, 2, and 5 μM for 48 h were examined by western blot using antibodies against total and phosphorylated AKT, NF-kB, JNK and ERK1/2 (Figure 2A and B). Following treatment, the protein levels of phospho-JNK increased, and those of phospho-AKT and phospho-NF-ĸB decreased. By contrast, the protein levels of total AKT, total NF-ĸB, total JNK, and total and phospho-ERK1/2 were not changed.
To document the roles of signal proteins in survival of cells exposed to 5 μM evodiamine for 48 h, cells were pretreated with the PI3K inhibitor wortmannin, the NF-ĸB inhibitor BAY11-7082, the JNK inhibitor SP600125 or the MEK inhibitor PD98059. After treatment, cell viability (Figure 2C) and cytotoxic activity (Figure 2D) were measured. Wortmannin, but not BAY11-7082, SP600125 and PD98059, decreased cell viability, and increased cytotoxic activity.
When cells were incubated with wortmannin prior to treatment with 5 μM evodiamine for 48 h, the protein levels of cleaved PARP increased, and those of phospho-AKT and Bcl2 decreased. The protein levels of total AKT and Bax in cells treated with both wortmannin and evodiamine were not affected, compared with those in cells treated with evodiamine alone (Figure 2E and F).
Evodiamine suppresses proliferation and migration of thyroid carcinoma cells. To identify the effect of evodiamine on cell proliferation, cells were treated with 5 μM evodiamine for 48 h, and then cell growth was evaluated by using cell number count (Figure 3A). Cell growth was reduced following treatment with evodiamine.


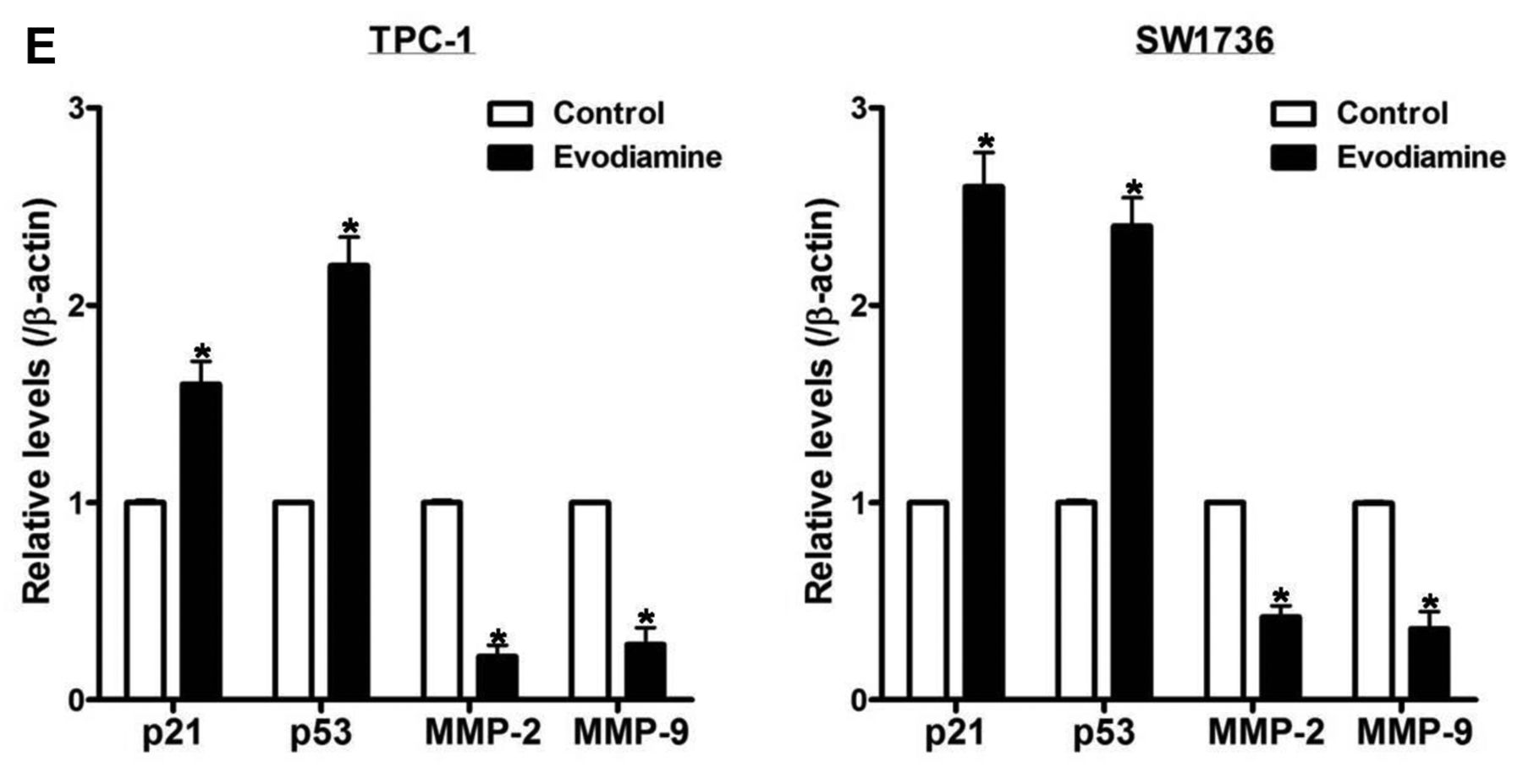
The impact of evodiamine on proliferation and migration of thyroid carcinoma cells. A: TPC-1 and SW1736 cells were treated with 5 μM evodiamine for 48 h, and cell growth was assayed using cell number count. B and C: TPC-1 and SW1736 cells were treated with 5 μM evodiamine for 48 h, and then cell migration was measured using wound healing assay. D and E: TPC-1 and SW1736 cells were treated with 5 μM evodiamine for 48 h, after which the protein levels of p21, p53, MMP-2 and MMP-9 were measured, and quantified by densitometry, and normalized to β-actin levels. All experiments were performed in triplicate. The blots are representative of three independent experiments. Data are expressed as mean±S.E. *p<0.05 vs. each matched control.
To inspect the influence of evodiamine on cell migration, cells were treated with 5 μM evodiamine for 48 h, and cell migration was estimated using wound healing assay (Figure 3B and C), and the protein levels of p21, p53, MMP-2 and MMP-9 (Figure 3D and E) were examined by western blot. After treatment, cell migration and the protein levels of MMP-2 and MMP-9 were reduced, while protein levels of p21 and p53 were elevated.
Evodiamine mitigates TGF-β-activated EMT in thyroid carcinoma cells. Evodiamine ameliorates cancer progression by alleviating migration, invasion and metastasis of cancer cells (5, 16). In this respect, evodiamine interrupts cell migration and invasion through modulation of MMP-2 and MMP-9 in prostate and nasopharyngeal cancer cells, and inhibits formation of metastatic lesions in colon cancer mice model (17-19). With regard to EMT, evodiamine attenuates HGF-induced invasiveness in melanoma, lung and colon cancer cells, and abrogates self-renewal via repression of EMT in gastric cancer stem cells (15, 20). Moreover, evodiamine impedes TGF-β-induced EMT in NRK52E cells and liver fibrosis in rats (21, 22). Thus, we examined whether evodiamine affects TGF-β-stimulated EMT in thyroid carcinoma cells.
To explore the impact of evodiamine on TGF-β-stimulated EMT, cells were treated with both 5 μM evodiamine and 100 ng/ml TGF-β for 48 h, and then cell growth (Figure 4A), morphology (Figure 4B) and migration (Figure 4C) were measured. Following treatment, TGF-β raised cell growth and migration, and resulted in fibroblast-like phenotype. Evodiamine abolished the TGF-β-induced alterations in cell growth, morphology and migration.
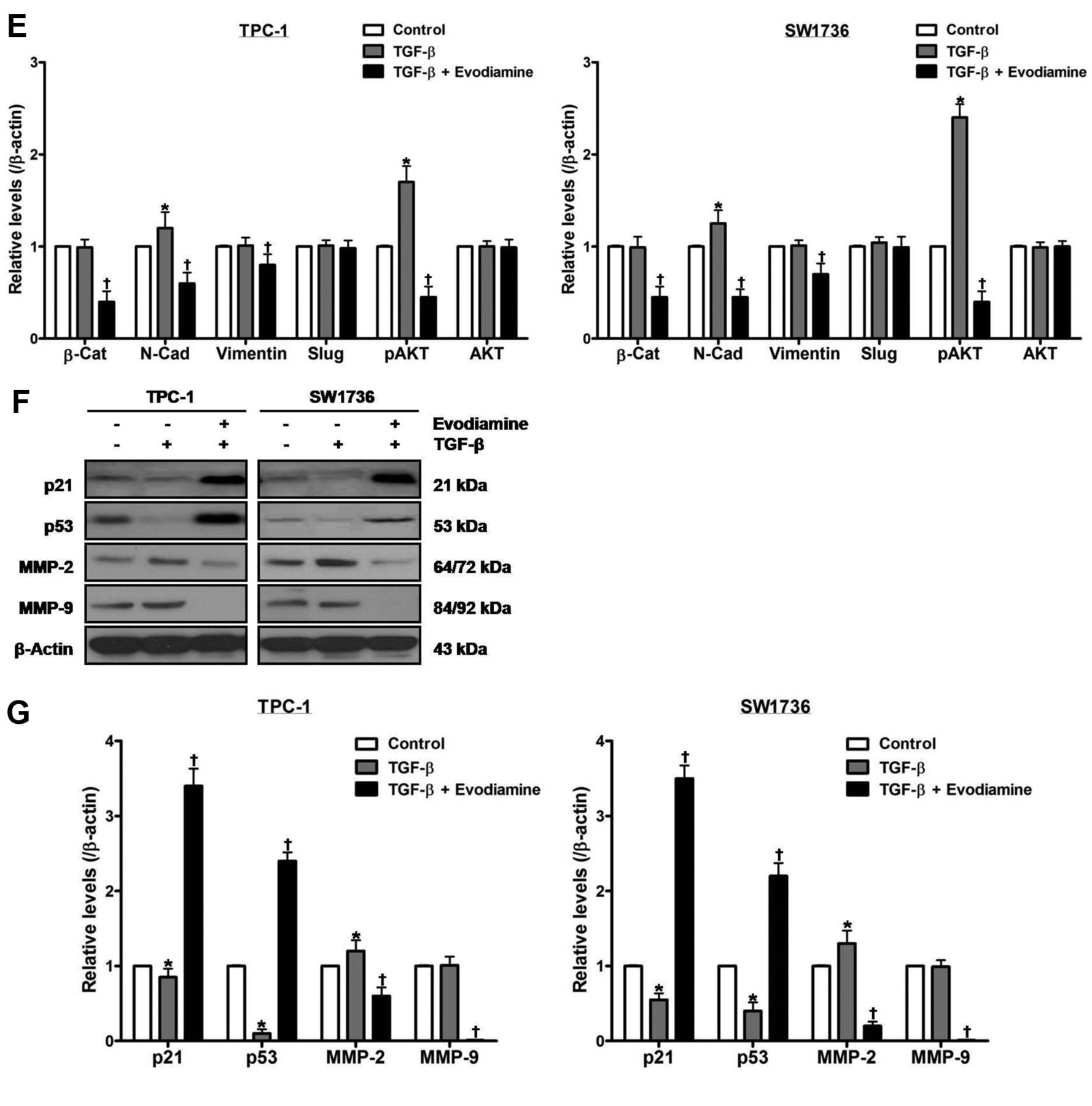
Treatment of cells with 100 ng/ml TGF-β for 48 h enhanced the protein levels of N-cadherin, phospho-AKT and MMP-2, and diminished those of p21 and p53 without affecting those of β-catenin, vimentin, slug, total AKT and MMP-9 (Figure 4D-G). Treatment of cells with both evodiamine and TGF-β, compared with TGF-β-treated cells, enhanced the protein levels of p21 and p53, and diminished those of β-catenin, N-cadherin, vimentin, phospho-AKT, MMP-2 and MMP-9 without affecting those of slug and total AKT.
Evodiamine synergizes with chemotherapeutic agents in exerting cytostatic and cytotoxic effects on thyroid carcinoma cells. To assess the cytostatic effect of the combination of evodiamine with chemotherapeutic agents, cells were treated with both evodiamine and doxorubicin, paclitaxel or cisplatin, and then the interactions were interpreted by obtaining CI using Chou-Talalay equation, where CI<1.0 indicates synergism, and CI=1.0 indicates additivity, and CI>1.0 indicates antagonism (Figure 5A and B, Table I). Cell growth was assayed by using cell number count, and the inhibition rate was computed as 100-cell growth (%). Following cotreatment, all CI values were lower than 1.0, and the combination data points were all located below the isobologram line at ED50, suggesting the synergism between evodiamine and chemotherapeutic agents in leading to cytostatic activity in thyroid carcinoma cells.

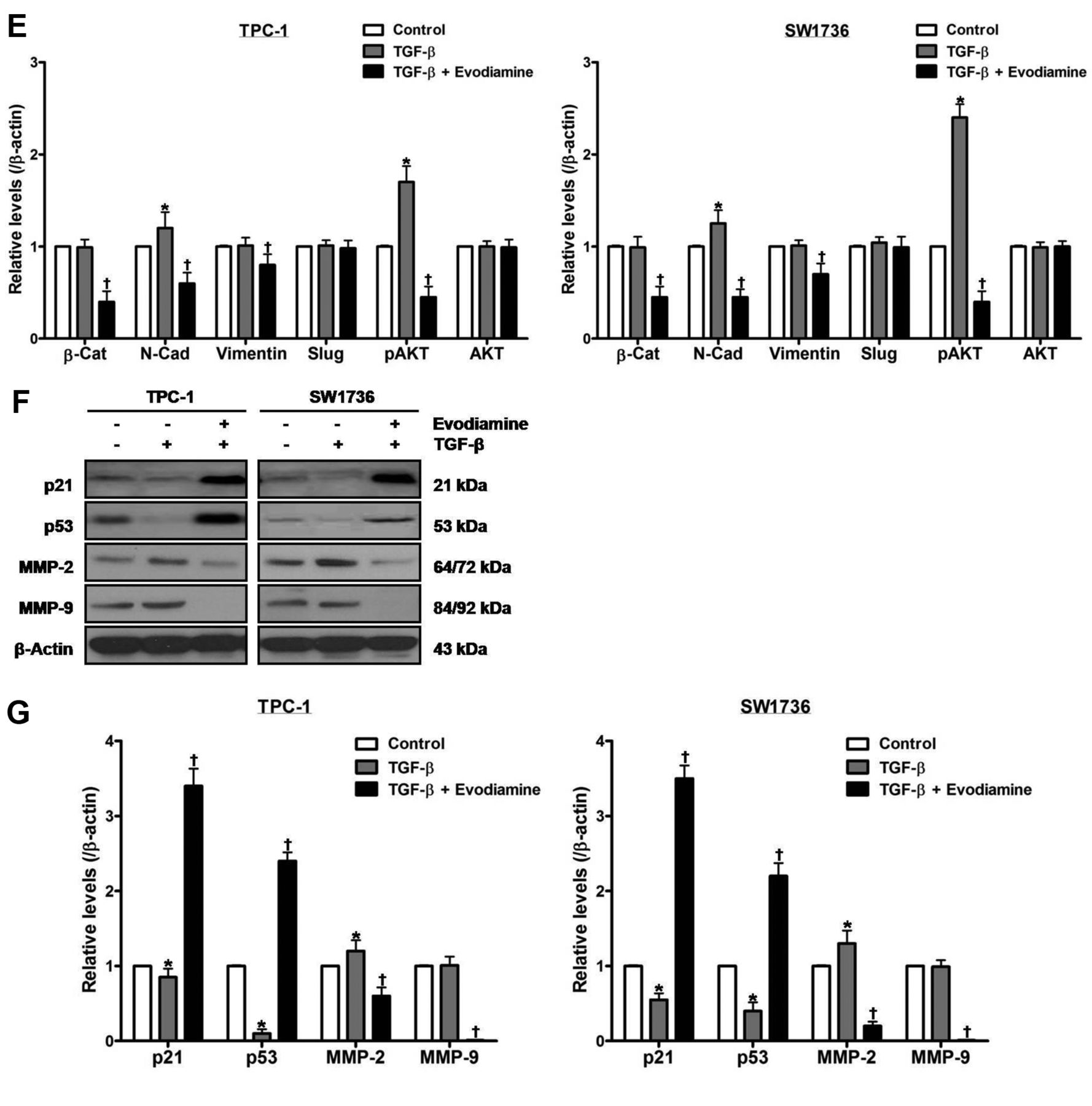
The effect of evodiamine on TGF-β-stimulated epithelial–mesenchymal transition in thyroid carcinoma cells. TPC-1 and SW1736 cells were treated with both 5 μM evodiamine and 100 ng/ml TGF-β for 48 h, and cell growth (A), morphology (B) and migration (C) were measured. D and E: TPC-1 and SW1736 cells were treated with both 5 μM evodiamine and 100 ng/ml TGF-β for 48 h, and then the protein levels of β-catenin, N-cadherin, vimentin, slug, and total and phospho-AKT were measured. F and G: TPC-1 and SW1736 cells were treated with both 5 μM evodiamine and 100 ng/ml TGF-β for 48 h, after which the protein levels of p21, p53, MMP-2 and MMP-9 were measured. All experiments were performed in triplicate. The blots are representative of three independent experiments. Data are expressed as mean±S.E. *p<0.05 vs. each matched control; †p<0.05 vs. cells treated with both evodiamine and TGF-β. β-Cat: β-Catenin; N-Cad: N-Cadherin.
Combination index values of growth of thyroid carcinoma cells at combined doses determined by the median effect analysis method.
Combination index values of survival of thyroid carcinoma cells at combined doses determined by the median effect analysis method.
Next, to analyze the cytotoxic effect of the combination of evodiamine with chemotherapeutic agents, cells were treated with both evodiamine and the above chemotherapeutic agents (Figure 5C and D, Table II). Cell viability was measured using CCK-8 assay, and the death rate was calculated as 100-cell viability (%). After cotreatment, all CI values were lower than 1.0, and the combination data points were all placed below the isobologram line at ED50, implying that evodiamine synergistically has a cytotoxic effect with chemotherapeutic agents in thyroid carcinoma cells.
Discussion
This study demonstrated, for the first time, that evodiamine resulted in cell death with concomitant variations in survival-related proteins, and inactivation of AKT potentiated the negative effect of evodiamine on survival of thyroid carcinoma cells. Additionally, evodiamine attenuated proliferation, migration and EMT by modulating EMT-related proteins, and displayed synergism with chemotherapeutic agents in repressing the growth and survival of thyroid carcinoma cells.
Evodiamine shows remarkable antimitogenic action in a range of cancers including hormone-sensitive prostate and breast cancers on the cellular level (5). As one of feasible mechanisms, it has been reported that evodiamine blocks cell cycle progression at G2/M phase in conjunction with stimulation of Cdc2/cyclin B complex (6, 7). Meanwhile, Bcl2 family proteins perform multiple crucial functions for cellular homeostasis such as survival and proliferation (26). In this regard, relative expression of the pro-survival protein Bcl2 and the anti-survival protein Bax, called the Bcl2/Bax switch, has an influence on the survival of cancer cells (27). With respect to impact of evodiamine on survival-related proteins including Bcl2 family proteins, evodiamine causes cell death through suppression of Bcl2, Bcl-xL, survivin, inhibitor of apoptosis protein, MMP-9, cyclin D1 and cyclooxygenase 2 in a variety of cancer cells (8). Moreover, it has been shown that evodiamine leads to cell death via Bcl2 regulated by ubiquitin-proteasome system, Bcl2/Bax ratio, p21 and p53 in A375-S2 melanoma cells (9, 10). In HepG2 hepatoma cells, evodiamine results in cell death by reducing Bcl2/Bax ratio (11). Although it was presented that evodiamine inhibited survival and proliferation of ARO cells, thought to be ATC cells, the cells have been identified as colon cancer cells (7, 28). TPC-1 and SW1736 cells used in this study have been authenticated as PTC and ATC cells, respectively, and thus our findings are the first to report that evodiamine induces death of true thyroid carcinoma cells (28). Briefly, evodiamine reduced cell viability, and elevated cytotoxic activity and the percentage of apoptotic cells in a dose-dependent manner. Correspondingly, evodiamine reduced Bcl2 protein levels, whereas it did not change Bax protein levels, causing reduction of Bcl2/Bax ratio. In addition, evodiamine elevated the protein levels of γH2AX and cleaved PARP, and reduced those of survivin. These data indicate that evodiamine leads to cell death with concomitant alterations in expression of Bcl2, survivin and γH2AX in thyroid carcinoma cells. Furthermore, these results suggest that evodiamine exerts a cytotoxic activity through modulation of survival-related proteins in thyroid carcinoma cells. In this regard, further studies on clinical implications of evodiamine in thyroid cancer patients are necessary to examine whether cytotoxicity is reenacted in in vivo models.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The influence of the combination of evodiamine with chemotherapeutic agents on growth and survival of thyroid carcinoma cells. TPC-1 and SW1736 cells were treated with both 0.5, 1, 1.5, 2 μM evodiamine and 0.5, 1, 1.5, 2 μM doxorubicin, 0.5, 1, 1.5, 2 nM paclitaxel or 50, 100, 150, 200 μM cisplatin for 24 h. A and B: Cell growth was measured assayed by using cell number count, and the inhibition rate was calculated as 100-cell growth (%). C and D: Cell viability was measured using CCK-8 assay, and the death rate was computed as 100-cell viability (%). Combination index and isobologram were estimated. All experiments were performed in triplicate. Data are expressed as mean±S.E.
PI3K/AKT signaling orchestrates many intracellular processes essential for survival of healthy cells (29). In thyroid carcinoma cells, it has been shown that aberrant activation of PI3K/AKT signaling participates in neoplastic transformation, and PI3K/AKT signaling plays pivotal roles in cell survival in our previous reports (30-37). With respect to the involvement of PI3K/AKT signaling in evodiamine-induced antitumor action, it has been shown that evodiamine enhances the therapeutic efficacy of gemcitabine via direct or indirect negative regulation of PI3K/AKT signaling in in vitro and in vivo models of pancreatic cancer (12). Moreover, evidence has been provided clarifying that evodiamine results in cell death by diminishing phospho-AKT protein levels in HepG2 hepatoma cells, and represses cell proliferation through inactivation of PI3K/AKT signaling and PTEN in osteosarcoma cells (11, 13). In A375-S2 melanoma cells, evodiamine causes cell death via diminution of PI3K/AKT signaling and enhancement of Fas-L/NF-ĸB signaling, and deactivation of PI3K/protein kinase C and ERK cascade with inactivation of SIRT1 (9, 10). In addition to PI3K/AKT signaling, evodiamine has anticancer action by modulating different signaling pathways such as NF-ĸB, mitogen-activated protein kinases and Wnt/β-catenin (8, 14, 15). In the present study, evodiamine enhanced the protein levels of phospho-JNK, and diminished those of phospho-AKT and phospho-NF-ĸB without affecting those of total AKT, total NF-ĸB, total JNK, and total and phospho-ERK1/2. However, in cells treated with evodiamine, the PI3K inhibitor wortmannin only, but not the inhibitors of NF-ĸB, JNK and ERK1/2, diminished cell viability, and enhanced cytotoxic activity. Furthermore, wortmannin enhanced the protein levels of cleaved PARP, and diminished those of phospho-AKT and Bcl2 without affecting those of total AKT and Bax. These findings manifest that suppression of AKT stimulates cell death induced by evodiamine in thyroid carcinoma cells. Considering our previous studies verifying that inhibition of AKT intensifies the cytotoxic activity of various agents in thyroid carcinoma cells (38-40), the results of this study imply that repression of AKT reinforces evodiamine-induced cytotoxicity in thyroid carcinoma cells.
The p21, p53 and matrix metalloproteinases are related to proliferation, migration, invasion and metastasis of cancer cells (41-43). In this regard, we have previously reported that p21, p53, MMP-2 and MMP-9 are responsible for proliferation and migration of thyroid carcinoma cells (37, 44). Besides its cytostatic and cytotoxic effects, evodiamine impedes cancer progression through interruption of migration, invasion and metastasis of cancer cells (5, 16). In in vitro and in vivo studies, it has been shown that evodiamine suppresses cell migration and invasion via MMP-2 and MMP-9 in prostate and nasopharyngeal cancer cells, and decreases metastatic foci in colon cancer cell-inoculated nude mice (17-19). With respect to EMT, evodiamine has been documented to inactivate bioactive HGF, and thereby inhibit HGF-stimulated invasiveness in melanoma, lung and colon cancer cells (20). Moreover, evodiamine lessens self-renewal by ameliorating EMT in a process of canonical Wnt/β-catenin signaling in gastric cancer stem cells, and exhibits a repressive property on TGF-β-stimulated EMT in NRK52E cells and liver fibrosis in rats (15, 21, 22). In the present study, evodiamine decreased cell growth and migration in conjunction with increment of the protein levels of p21 and p53 as well as decrement of those of MMP-2 and MMP-9. TGF-β accelerated cell growth and migration, and induced a fibroblast-like phenotype, while evodiamine abolished the TGF-β-induced changes in cell growth, morphology and migration. In TGF-β-treated cells, the protein levels of N-cadherin, phospho-AKT and MMP-2 increased, and those of p21 and p53 decreased but those of β-catenin, vimentin, slug, total AKT and MMP-9 were not altered. In cells treated with both evodiamine and TGF-β, compared with TGF-β alone, the protein levels of p21 and p53 increased, and those of β-catenin, N-cadherin, vimentin, phospho-AKT, MMP-2 and MMP-9 decreased, whereas those of slug and total AKT were unchanged. Taken together, these data reveal that evodiamine mitigates cell proliferation and migration with concomitant overexpression of p21 and p53 as well as underexpression of β-catenin, N-cadherin, vimentin, phospho-AKT, MMP-2 and MMP-9 in TGF-β-treated thyroid carcinoma cells. In addition, these results suggest that evodiamine negatively affects TGF-β-induced EMT of thyroid carcinoma cells through regulation of EMT-related proteins.
In the national guidelines, it is recommended that chemotherapeutic agents may be considered in tyrosine kinase inhibitor-refractory WDTC and ATC patients (2, 3). In this regard, doxorubicin has been approved as monotherapy, but its clinical efficacy and response are not satisfactory in ATC patients (45). Furthermore, it has been reported that doxorubicin fails to eliminate cancer stem cells derived from ATC cells (46). With respect to other chemotherapeutic agents, paclitaxel was shown to be a more beneficial chemotherapeutic agent than those traditionally used in ATC patients but we have reported that the heat shock protein 90 inhibitor 17-allylamino-17-demethoxygeldanamycin antagonizes with paclitaxel in inducing death of ATC cells (32, 47). Moreover, cisplatin alone or in combination with docetaxel was shown to have an influence on ATC patients (3, 48). In view of the combination of evodiamine with chemotherapeutic agents, evodiamine induces cell death in chemoresistant breast cancer in vitro and in vivo models (23, 24). In addition, evodiamine leads to cytostatic and cytotoxic action in A2780 ovarian cancer cells refractory to paclitaxel (25). In the present study, when evodiamine was combined with the chemotherapeutic agents doxorubicin, paclitaxel or cisplatin, all CI values were lower than 1.0 in terms of inhibition and death rates. These findings demonstrate that evodiamine has a synergistic activity with doxorubicin, paclitaxel or cisplatin in suppressing growth and survival of thyroid carcinoma cells. Furthermore, these results imply that evodiamine acts synergistically with chemotherapeutic agents to induce cytostasis and cytotoxicity in thyroid carcinoma cells. Therefore, combination of evodiamine with chemotherapeutic agents may be an attractive therapeutic regimen in human thyroid cancer.
In conclusion, our results suggest that evodiamine poses antioncogenic action by modulating survival-related proteins, and inhibition of AKT magnifies evodiamine-induced cytotoxic activity in thyroid carcinoma cells. Moreover, evodiamine abrogates proliferation, migration and EMT by regulating EMT-related proteins, and synergizes with chemotherapeutic agents in inducing cytostasis and cytotoxicity in thyroid carcinoma cells. Although our data should be scrutinized in an in vivo model, this study implies that evodiamine alone or in combination with chemotherapeutic agents is a promising remedy in thyroid cancer patients resistant to conventional approaches.
Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIP) (No. 2018R1D1A1B07044901) to S.J. Lee, Republic of Korea, and was also supported by Hallym University Research Fund to S.J. Lee, Republic of Korea.
- Received October 13, 2018.
- Revision received October 24, 2018.
- Accepted October 25, 2018.
- Copyright© 2018, International Institute of Anticancer Research (Dr. George J. Delinasios), All rights reserved