

Abstract
Background: Malignant mesothelioma (MM) is a well-known malignant tumor that occurs in the pleura and is histopathologically classified into three subtypes. Lymphohistiocytoid mesothelioma (LHM) is considered a variant of epithelioid MM, and few cases have been reported. First case of LHM was reported by Henderson et al. in 1988. It is difficult to precisely diagnose LHM, and it is often misdiagnosed as reactive mesothelial cell proliferation. Case report: An 82-year-old man, with the smoking history of nine pack-years, was referred to our Department due to an abnormal shadow and pleural effusion in the left lung field on the chest X-ray imaging. His occupation was a teacher through his life without any asbestos exposure. Computed tomography (CT) and 18F-fluorodeoxyglucose-position emission tomography showed a tumor which was suggestive of malignancy on the left chest wall, with the possible invasion into the left 2nd to 4th ribs. He underwent a CT-guided biopsy and a thoracentesis, but the tumor was shown to be a benign tumor indicative of a reactive mesothelial cell proliferation. Then, he underwent a surgical resection and the tumor was suspected of liposarcoma macroscopically. Histological and immunohistochemical findings were suggestive of mesothelial lesion, such as nodular histiocytic or mesothelial hyperplasia. However, loss of BAP1 and no p16 homozygous deletion in the tumor cells led to the diagnosis of LHM, not a benign lesion.
- Malignant mesothelioma
- lymphohistiocytoid mesothelioma
- immunohistochemistry
- BAP1
Malignant mesothelioma (MM) is a common pleural neoplasm mainly caused by asbestos exposure, and its prognosis is poor, although multimodality treatment, including surgery, chemotherapy and radiotherapy, is administered for patients with MM (1). MM arises in cells lining the serosal membranes surface, mainly pleural but also peritoneal or pericardial (2). MM has a wide histopathologic spectrum of various cell types, and it can be classified as three typical cell types, which are epithelioid, sarcomatoid, and biphasic consisting of both epithelioid and sarcomatoid components (3). Diagnostic methods include cytological and histological examination. A recent study demonstrated the usefulness of various kinds of diagnostic methods in diagnosing MM. For instance, Hida et al. showed that the combination of BRCA-1-associated protein 1 (BAP1) immunohistochemistry (IHC) and p16 fluorescence in situ hybridization (FISH) assays is highly sensitive in differentiating MM from reactive mesothelial proliferation (4).
Lymphohistiocytoid mesothelioma (LHM) is regarded as a very rare variant of epithelioid MM, and the first case was described by Henderson et al. in 1988 (5). Previously, LHM was originally classified as sarcomatoid MM; however, literature reported that patients diagnosed with this subtype showed a similar prognosis as that of epithelioid subtype: Kawai T et al. showed that the median overall survival of 36 cases with LHM was as long as 18.2 months (6). Thus, LHM was currently classified as epithelioid MM. The frequency of LHM is less than 1% of MM, although it is 3.3% due to the report by Yao et al. (7). LHM is difficult to diagnose accurately, and should be mainly differentiated from Hodgkin and non-Hodgkin malignant lymphoma, lymphoepithelial carcinoma, thymic tumors, and reactive mesothelial cell proliferation (8). In particular, a reactive mesothelial cell proliferation, which is a benign lesion, often mimics MM histologically and cytologically (9, 10). Thus, it is important to accurately diagnose LHM.
Here, we report a rapidly progressive case which had been considered a reactive mesothelial proliferation firstly, but was exactly diagnosed as LHM by using BAP1 IHC.

The radiological images of the current case. The tumor shadow (circle) and the dull cost-phrenic angle which indicated the presence of pleural effusions were found in the left lung field on a chest X-ray (a). Computed tomography revealed a chest wall mass, approximately 58 mm, which appeared to invade the chest wall (b). 18F-fluorodeoxyglucose (FDG)-position emission tomography showed that the tumor exhibited avid intake of FDG (c).

Thoracoscopic findings showed that the chest wall tumor (arrow head) which adhered to or invade the left lingual segment (a). The resected tumor contained a fatty-like tissue (b).
Case Report
An 82-year-old male visited hospital regularly because of hypertension and gout. He was an ex-smoker with a history of 10 cigarettes per day from the age of 22- to 40-years-old. His life-time occupation is a teacher without any definite exposure to asbestos. Although no symptoms were observed, a chest X-ray imaging pointed out the abnormal shadow and a little amount of pleural effusion in the left lung field on a Chest X-ray (Figure 1a). Laboratory test showed no abnormality, including inflammatory markers. Computed tomography (CT) showed a chest wall mass shadow measuring approximately 58 mm, which was considered to invade the dorsal side of the 2nd to 4th ribs (Figure 1b). Moderate amount of pleural effusion was observed, and no lymphadenopathy was seen. 18F-fluorodeoxyglucose-position emission tomography (FDG-PET) showed avid intake of FDG, with the maximum standardized uptake value being 8.23 (Figure 1c), suggesting the possibility that the mass shadow might be a malignant tumor.
He underwent a CT guided biopsy which showed fragments of desquamated mildly atypical, epithelioid and swollen mesothelial cells, which were immunohistochemically positive for calretinin, arranged in sheets or cords, admixed with an inflammatory exudate containing CD68-immunoreactive macrophages. In addition, mitotic figures of mesothelial cells were not seen. The feature was suggestive of reactive mesothelial cell proliferation. However, since further scrutiny was required because MM could not be denied, he was referred to our department.

{kind=link}
{kind=link}
{kind=link}
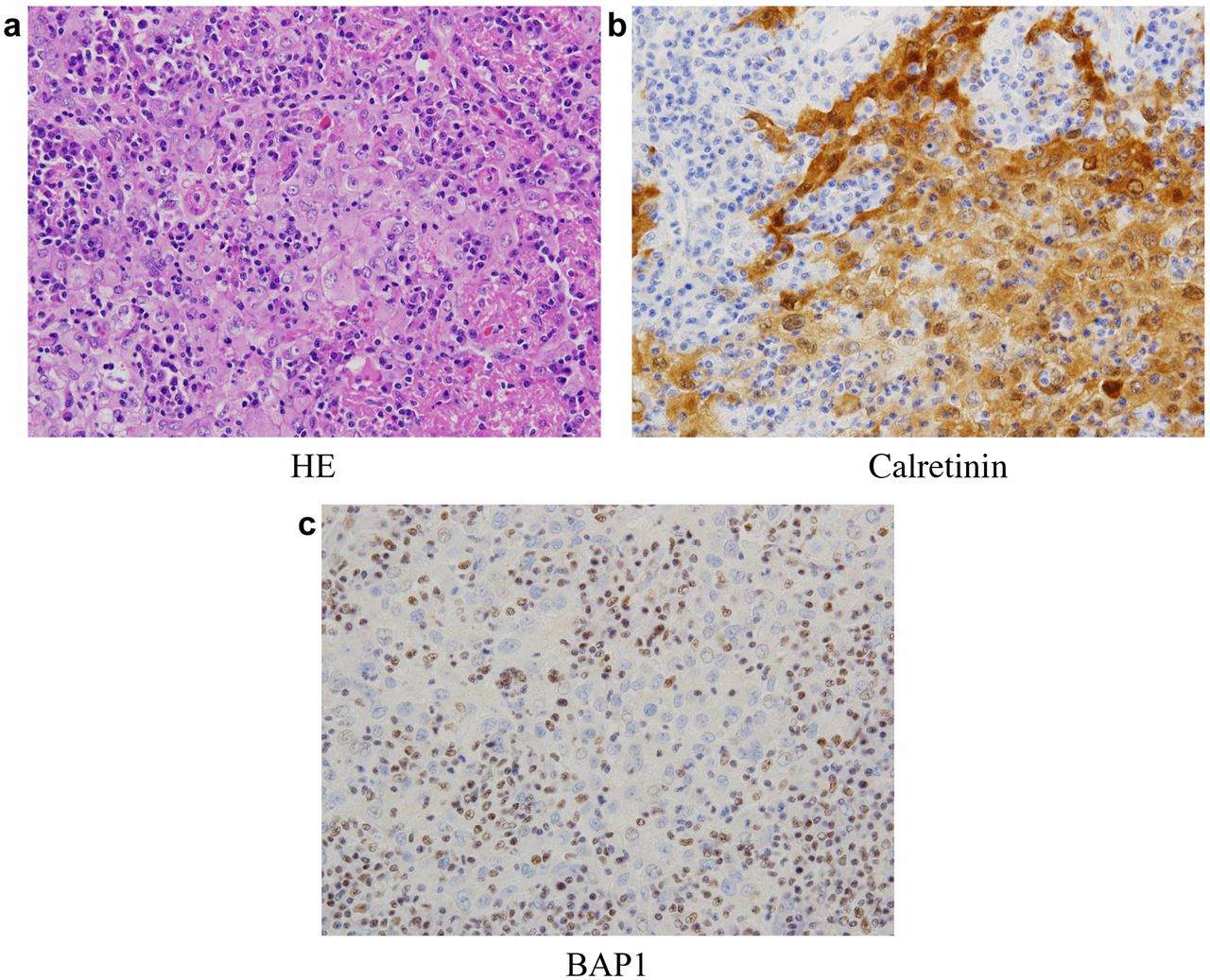
The tumor showed a proliferation of atypical mesothelial cells with prominent lymphohistiocytic infiltration (HE). Immunohistochemistry for calretinin and BAP1 showed the immuno-positivity for calretinin and the loss of BAP1 protein expression.
First, the patient underwent a thoracentesis and a bloody fluid was found. The cytological examination revealed that the fluid contained the scattered neutrophilic leukocytes, and no malignant tissues. Subsequent CT scan showed that the tumor measured 72 mm in size, although 14 mm about two months before, suggesting that the tumor might be a malignant tumor. Therefore, surgical resection was planned for the exact diagnosis and treatment.
Surgical findings. A video-assisted thoracoscopic surgery was performed. A pleural tumor, measuring approximately 80 mm in size, was considered to invade the 2nd intercostal space, 3rd/4th ribs, and 4th intercostal space. In addition, adhesion was observed between the tumor and left lingual segment (Figure 2a). Moderate amount of bloody pleural effusion was observed, and cytological examination revealed no malignant cells. Therefore, we performed tumor extirpation with the combined resection of 3rd/4th ribs and partial resection of left lingual segment. The tumor macroscopically contained fatty-like tissues (Figure 2b), and therefore, we suspected the tumor of liposarcoma. The patient was discharged from hospital on eight postoperative days without any complications, and he is doing well 8 months after the operation.
Pathological findings. Histologically (Figure 3a), the tumor was composed of polygonal or epithelioid cells with enlarged oval nuclei, conspicuous nucleoli and eosinophilic cytoplasm arranged in a sheet-like or cord-like pattern, accompanied by lymphohistiocytic infiltration. Occasional mitotic figures and necrosis were also associated. Immunohistochemically, the tumor cells were positive for pan-cytokeratin (AE1/AE3), calretinin (Figure 3b), D2-40, EMA and GLUT1, but negative for WT-1, IMP3, CD146, TTF-1, CEA, claudin-4, p40, S-100 protein and c-kit. In addition, loss of BAP1 was detected in the tumor cells (Figure 3c). No p16 homozygous deletion was demonstrated by fluorescence in situ hybridization (FISH). From the above feature, the tumor was diagnosed as a malignant mesothelioma, specifically, lymphohistiocytoid mesothelioma.
Discussion
MM, which is known as a rare pleural neoplasm deeply associated with asbestos exposure, is a highly malignant neoplasm, whose mortality is increasing in the developed countries, including Japan. The MM is originated from the mesothelial cell on the surface of the serosal membranes (2). It occurs mainly in pleural mesothelial cells, whose incidence is over 65%; however, sometimes peritoneal and pericardial can be involved, with the incidence being less than 10%. Furthermore, the 2015 World Health Organization Classification of Tumor of the Pleura classified MM into three types: epithelioid, sarcomatoid, and biphasic (3). Within the category of pleural epithelioid MM, a variety of morphologic subtypes are recognized, i.e., tubulopapillary, papillary, micropapillary, trabecular, solid, and lymphohistiocytoid.
Lymphohistiocytoid mesothelioma (LHM) is a very rare subtype of epithelioid MM, which comprised less than 1% of all subtypes of diffuse MM reported in the past literature: 0.8% (5), 0.5% (11), and 0.96% (12). In contrast, the incidence was 3.3% due to the report by Yao et al., whose study included cases diagnosed only by electron microscopic study. LHM was categorized as sarcomatoid MM, with an associated dense lymphocytic infiltration (13). However, the survival analysis of patients with LHM performed by Galateau-Salle et al. demonstrated the prognosis similar to that of epithelioid subtype and better than biphasic and sarcomatoid (12). In this case, the progression was very rapid and the tumor was likely to be a poorly differentiated neoplasm; however, the final pathological diagnosis was LHM. Eight months after the surgical resection, he is doing well without recurrence.
The present case had been diagnosed as reactive mesothelial cell proliferation at first by CT guided-biopsy. To diagnose LHM accurately, the histological differential diagnosis should be considered, such as Hodgkin and non-Hodgkin malignant lymphoma, lymphoepithelial carcinoma, sarcomatoid carcinoma of the lung, thymic tumor, poorly differentiated sarcoma, and reactive mesothelial cell proliferation (10, 12). Among them, reactive mesothelial cell proliferation often mimics MM histologically and cytologically (9, 10). Invasion of the mesothelial cell into the stromal or fat tissue is an essential and characteristic histological finding of MM (9); however, it is not always conspicuous, particularly in small biopsy specimens. To differentiate MM from others, IHC for various markers is useful. Some literatures described that BAP1 IHC assay is valuable for the differentiation of MM from reactive mesothelial cell proliferation (4, 14-15). Hida T et al. reported that 27 of 40 (67.5%) MM cases had loss of BAP1, and all 20 cases of reactive mesothelial proliferations did not have loss of BAP1, with its sensitivity and specificity being 67.5% and 100%, respectively (4). This patient, whose clinical course suspected MM, was firstly diagnosed as reactive mesothelial cell proliferation in biopsy and thoracentesis cytological findings. However, BAP1 IHC using surgical specimen suggested that the tumor was not reactive mesothelial cell proliferation, but LHM because of loss of BAP1. Thus, BAP1 is considered a key diagnostic tool which helps differentiating LHM from other diseases, specifically reactive mesothelial cell proliferation.
In conclusion, we reported a rapidly progressive LHM case, in which BAP1 was useful for the exact diagnosis, emphasizing the BAP1 IHC in differentiating LHM from reactive mesothelial cell proliferation.
Acknowledgements
The Authors would like to thank the Edanz Group (Fukuoka, Japan) for their assistance with English language revision.
Footnotes
Conflicts of Interest
The Authors have no competing interests.
- Received September 18, 2017.
- Revision received September 30, 2017.
- Accepted October 2, 2017.
- Copyright© 2017, International Institute of Anticancer Research (Dr. George J. Delinasios), All rights reserved