

Abstract
Background/Aim: The monopolar spindle 1 (MPS1) is a serine/threonine kinase that plays an important role in spindle assembly checkpoint signaling. To determine the possible relationship between MPS1 inhibition and genotoxic stress responses, herein we examined whether MPS1 inhibition influences cellular susceptibility towards two genotoxic treatments, etoposide and ionizing radiation (IR). Materials and Methods: Two murine tumour cell lines, SCCVII and EMT6, were used. The effect of genotoxic treatments with or without two novel MPS1 inhibitors, NMS-P715 and AZ3146, on cellular survival, cell-cycle distribution, centrosome status and mitotic catastrophe (MC) was evaluated. Results: MPS1 inhibition sensitized murine tumour cells to etoposide but not to IR. In addition, MPS1 inhibition altered cell-cycle progression and exacerbated centrosome abnormalities, resulting in enhanced MC induced by etoposide but not by IR. Conclusion: MPS1 inhibition promotes the etoposide-induced aberrant mitosis and, consequently, the induction of tumour cell death.
- MPS1
- etoposide
- cell death
- centrosome amplification
- mitotic catastrophe
In eukaryotes, the spindle assembly checkpoint (SAC) maintains genome integrity by delaying cell division until correct chromosome segregation is ensured during mitosis. Monopolar spindle 1 (MPS1, also known as TTK) is an evolutionarily conserved serine/threonine kinase that plays an important role in SAC signaling. Although MPS1 plays multiple roles in mitosis, the most widely conserved and prominent function is to regulate SAC activity by recruiting various SAC-related proteins to the kinetochore (1, 2). Furthermore, MPS1 also regulates chromosome alignment during metaphase, maintains chromosome integrity and controls centrosome duplication (3-5). Therefore, MPS1 is considered essential for the proper progression of mitosis (6).
Similar to other cell cycle-related proteins, MPS1 is overexpressed in several human cancers, including thyroid papillary carcinoma, breast cancer and lung cancer (7-10). MPS1 overexpression in a patient's tumour is positively correlated with tumour aggressiveness and poor survival (11, 12). These findings suggest that MPS1 has a role in cellular proliferative activities and tumour malignancy highlighting MPS1 as a potential therapeutic target for cancer therapy. Accordingly, several small-molecule MPS1 inhibitors have been developed; some of which have demonstrated potent cytotoxicity in various tumour cell lines (12-16).
In pre-clinical and clinical studies, the combination of a chemotherapeutic agent with a cell cycle-targeting drug is a strategy showing promise as cancer treatment. For example, Chk1 inhibition abrogates the DNA damage-induced G2 checkpoint and enhances cell death (17). This result suggests that inhibiting proteins involved in checkpoint regulation may sensitize cells to genotoxic treatments. In recent studies, MPS1 inhibitors (Mps-BAY2b and MPS-IN-3) have been shown to increase the sensitivity to a microtubule poison (paclitaxel and vincristine) in both human colon cancer and glioblastoma cells (12, 18). These results indicate that targeting MPS1 could be a potential novel approach in combination therapy. However, to our knowledge, only a few studies have tested this strategy with genotoxic stress. Recently, two novel MPS1 inhibitors, NMS-P715 and AZ3146, were developed based on their competitive mechanism against the MPS1 ATP-binding domain (1, 13). To determine the possible relationship between MPS1 inhibition and genotoxic stress responses, herein we examined whether MPS1 inhibition influences cellular susceptibility towards two genotoxic treatments, etoposide and ionizing radiation (IR) in murine tumour cells using NMS-P715 and AZ3146.
Materials and Methods
Reagents. Etoposide and nocodazole were purchased from Wako Pure Chemical Industries (Osaka, Japan). 4’, 6-Diamidino-2-phenylindole (DAPI) was purchased from Life Technologies (Carlsbad, CA, USA). NMS-P715 and AZ3146 were obtained from Merck (Kenilworth, NJ, USA) and AdooQ Bioscience (Irvine, CA, USA), respectively. The following antibodies were used: anti-γ-tubulin (Sigma-Aldrich, St. Louis, MO, USA), anti-phospho-histone H3 (Ser10) (Cell Signaling Technology, Beverly, MA, USA) and Alexa Fluor® 488 anti-mouse IgG (Life Technologies).
Cell culture and X-irradiation. Murine squamous carcinoma SCCVII cells were maintained in α-MEM medium (Life Technologies) supplemented with 10% (v/v) fetal bovine serum (FBS) (Biowest; Nuaille, France) at 37°C in a humidified atmosphere of 5% CO2. Murine breast cancer EMT6 cells were maintained in RPMI 1640 medium (Life Technologies) supplemented with 10% (v/v) FBS at 37°C in a humidified atmosphere of 5% CO2. X-irradiation was performed with a Shimadzu PANTAK HF-320 X-ray generator (Shimadzu, Kyoto, Japan) at 200 kVp, 20 mA with a 1.0-mm aluminium filter or an X-Rad iR-225 (Precision X-Ray, North Branford, CT, USA) at 200 kVp, 15 mA with a 1.0-mm aluminium filter.
Phospho-histone H3 staining. Cells were treated with nocodazole (100 ng/ml) for 6 h, followed by the addition of MPS1 inhibitors at the indicated concentrations. After incubation for 2 h, the cells were collected and washed with ice-cold phosphate-buffered saline (PBS). The cells were then fixed with ice-cold 70% (v/v) ethanol and kept at −20°C for 12 h. After centrifugation, the cells were permeabilized with 0.25% (v/v) Triton X-100/PBS for 15 min at 4°C and incubated with PBS containing anti-phospho-histone H3 (Ser 10) antibody (1:50 dilution) for 3 h at room temperature (RT) on a rotator. After three washes with PBS, the cells were incubated in the dark with the Alexa Fluor 488-conjugated anti-mouse IgG (1:200 dilution) for 90 min and, subsequently, washed with PBS. Flow cytometry analysis was conducted using an EPICS XL™ flow cytometer (Beckman Coulter, Fullerton, CA, USA) and the population of p-H3-positive cells was determined.
Clonogenic survival assay. Cells were collected and seeded into 60-mm dishes at densities of 100-25,000 cells/dish and allowed to adhere for 6 h. For drug treatment, the cells were treated with etoposide in the presence or absence of the MPS1 inhibitors for 24 h. For X-irradiation, the cells were X-irradiated and then incubated with MPS1 inhibitors for 24 h. After treatment, they were washed twice with PBS, followed by the addition of fresh medium. The cells were allowed to proliferate in a humidified 5% CO2 atmosphere at 37°C for 6 d before being fixed with methanol and stained with Giemsa solution (Wako). Colonies containing >50 cells were scored as surviving cells. Surviving fractions were calculated with a correction for the plating efficiency of the cells treated with the MPS1 inhibitors.
Cell-cycle analysis. The cells were exposed to etoposide or X-rays, with or without the MPS1 inhibitors, as described above. After incubation, the cells were collected and washed with ice-cold PBS. The cells were fixed with ice-cold 70% (v/v) ethanol and kept at −20°C for 12 h. RNA was hydrolyzed with 100 μg/ml RNase A (NIPPON GENE; Tokyo, Japan) at 37°C for 30 min and, subsequently, stained with propidium iodide (Sigma-Aldrich) for 20 min. The DNA content was measured using an EPICS XL flow cytometer.
Analysis of mitotic catastrophe (MC) and centrosome numbers. Analysis of MC and centrosome numbers was performed as previously described (19). Cells were seeded on glass coverslips coated with collagen (Cellmatrix Type I-C; Nitta Gelatin, Osaka, Japan) and cultured overnight. After treatment, the cells were fixed with ice-cold methanol for 10 min at −20°C. After permeabilization with ice-cold acetone for 1 min at −20°C, the cells were washed 3 times with PBS, followed by PBS treatment containing 6% (v/v) goat serum (Chemicon International, Temecula, CA, USA) for 1 h at RT. Subsequently, they were incubated with anti-γ-tubulin antibody (1:5,000 dilution) in 3% (v/v) goat serum/PBS overnight at 4°C. They were then incubated in the dark with the Alexa Flour 488-conjugated anti-mouse IgG (1:2,000 dilution) in 3% (v/v) goat serum/PBS for 1.5 h. After incubation, they were washed 3 times with PBS and counterstained with 300 nM DAPI for 5 min at RT. The coverslips were mounted with ProLong® Gold Antifade Mountant reagent (Life Technologies). Fluorescence microscopy analysis was performed using an Olympus BX61 microscope (Olympus, Tokyo, Japan) with a reflected light fluorescence. For MC analysis, at least 100 cells were counted; cells with features of aberrant mitotic nuclei (micro-, multilobular and fragmented nuclei) were scored as cells undergoing MC. For centrosome analysis, the number of γ-tubulin foci per cell was counted manually. At least 100 cells were analysed and the percentage of the cells containing more than two foci was determined.
Statistical analysis. All results are expressed as means±standard deviation (S.D.) of at least three separate experiments. Comparison of two groups was performed using Student's t-test. For multiple comparisons, Dunnett's test was performed. The minimum level of significance was set at p<0.05.
Results
Efficacy of MPS1 inhibitors to inactivate SAC. MPS1 inhibition is expected to inactivate SAC and override the mitotic arrest induced by spindle poisons, such as nocodazole (1, 13). To assess the potency of two novel MPS1 inhibitors, NMS-P715 and AZ3146, to evade SAC at the cellular level, we treated nocodazole-arrested SCCVII cells with these drugs at different concentrations. Cells undergoing mitotic arrest were identified by phosphorylated histone H3 (p-H3) staining. Although the proportion of p-H3-positive cells was 4% in untreated cells, it increased to 70% after nocodazole treatment (Figure 1a). When cells were treated with NMS-P715, the proportion of cells under mitotic arrest was reduced in a concentration-dependent manner up to 1 μM (Figure 1b). Similarly, AZ3146 decreased the proportion of mitotic-arrested cells in a concentration-dependent manner up to 1 μM (Figure 1c). These results demonstrate that both MPS1 inhibitors effectively inactivate SAC and override mitotic arrest.
Effects of MPS1 inhibitors on cellular sensitivity to genotoxic treatments. Next, we examined the cytotoxicity of the MPS1 inhibitors. We treated SCCVII cells with different concentrations of NMS-P715 or AZ3146 for 24 h and found that both MPS1 inhibitors were cytotoxic at concentrations of 0.6 μM or higher (Figure 2a and 2b). Moreover, NMS-P715 exhibited similar cytotoxic effects in another mouse tumour cell line, EMT6 (Figure 2c). These results show that the MPS1 inhibitors under investigation reduce cell viability in a concentration-dependent manner. In the following experiments, a subtoxic concentration (0.5 μM) was used to evaluate the effects of each MPS1 inhibitor in combination with etoposide or IR.
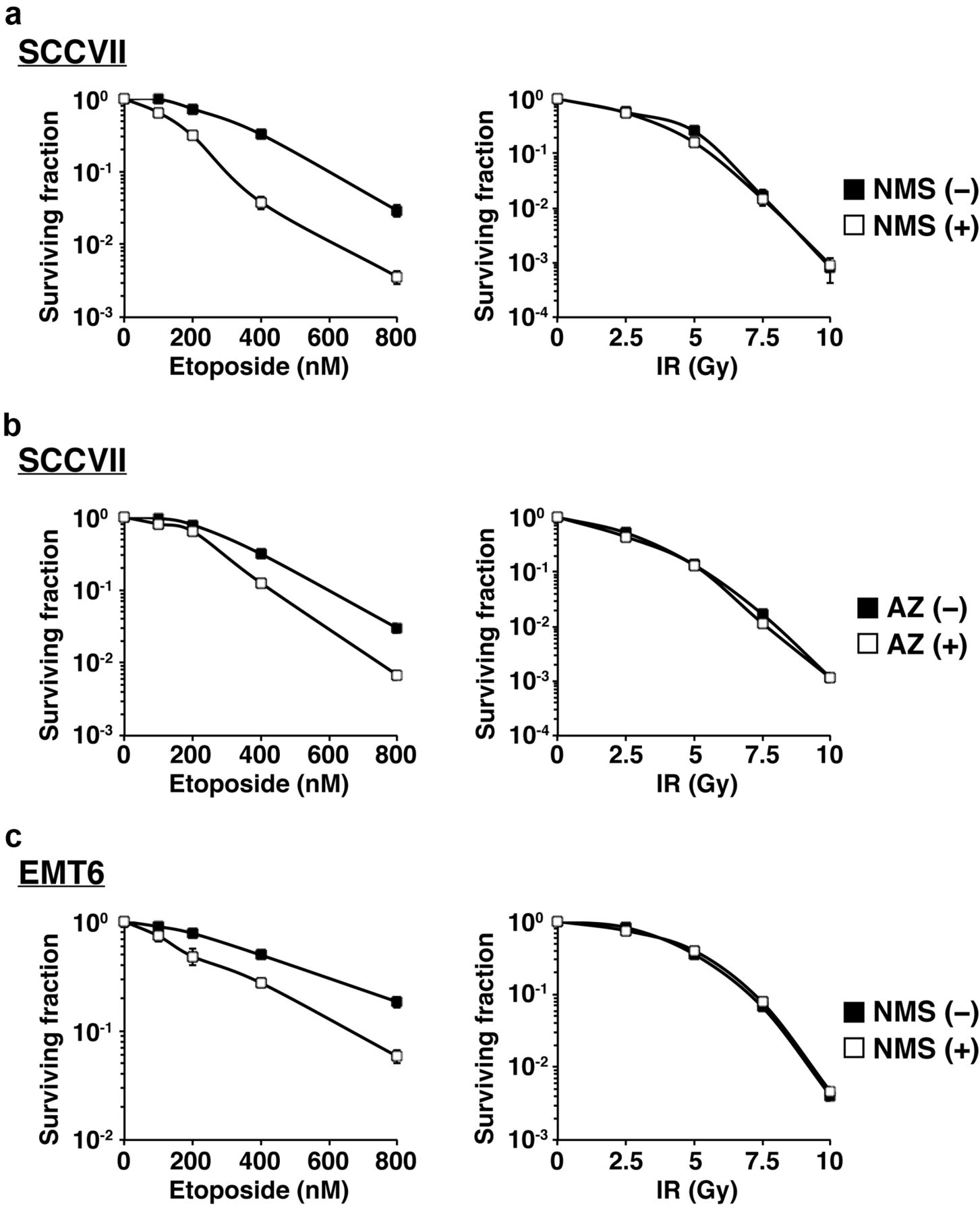
To determine whether MPS1 inhibition influences cellular sensitivity to genotoxic agents, we examined cell viability after treatment with etoposide or IR, with or without the MPS1 inhibitors, using a clonogenic survival assay. Etoposide treatment induced cytotoxicity in a concentration-dependent manner in SCCVII cells (Figure 3a; left). The addition of NMS-P715 significantly decreased cellular survival after etoposide treatment suggesting that NMS-P715 enhances cellular sensitivity to etoposide in SCCVII cells. AZ3146 also enhanced cellular sensitivity to etoposide in SCCVII cells (Figure 3b; left). A similar chemosensitizing effect was observed with NMS-P715 in EMT6 cells exposed to etoposide (Figure 3c; left). However, both MPS1 inhibitors had no impact on cellular radiosensitivity in either SCCVII or EMT6 cells (Figure 3a-c; right). These findings demonstrate that MPS1 inhibition sensitizes murine tumour cells to etoposide but not to IR.
Effect of MPS1 inhibitors on cell-cycle distribution after genotoxic treatments. To gain insight into the mechanism through which MPS1 inhibition enhances cellular susceptibility to etoposide, but not to IR, we analyzed the cell-cycle distribution of cells treated with etoposide or IR, with or without MPS1 inhibitors. Figure 4a shows the representative cell cycle profile of SCCVII cells after treatment with etoposide (400 nM) or IR (5 Gy) in the absence of MPS1 inhibitors. Etoposide promoted a distinct change in cell-cycle distribution, thus reducing the cell population in the G1 phase with a concomitant increase in the cell population in the G2/M phase, whereas, IR resulted in a similar, but weaker, cell-cycle change in SCCVII cells. These results indicate that, although both treatments induce G2/M arrest, etoposide is a more potent activator of cell-cycle checkpoints in this experimental setting. Quantitative analysis confirmed this finding not only in SCCVII cells but also in EMT6 cells (Figure 4b-d). Although treatment with an MPS1 inhibitor alone did not alter cell cycle profiles, co-treatment of the MPS1 inhibitors with etoposide resulted in a significant increase of the cell population in the G2/M phase in both SCCVII and EMT6 cells. In contrast, the MPS1 inhibitors did not alter the cell-cycle distribution after IR in either cell line. Therefore, MPS1 inhibition influenced the cell-cycle progression only in etoposide-treated cells but not in IR-treated cells. These results imply that these differences in cell-cycle progression may contribute to the differences in the sensitizing effects of the MPS1 inhibitors when treated with etoposide or IR.
Effect of MPS1 inhibitors on centrosome status after genotoxic treatments. Whereas a normal cell contains one or two centrosomes, the centrosome duplication system is often disturbed when cells are exposed to various stresses, thus leading to the formation of supernumerary centrosomes (20, 21). Supernumerary centrosome formation leads to aberrant mitosis, which generates daughter cells carrying aneuploidy and is associated with mitotic cell death (20, 21). Since the MPS1 inhibitors altered cell-cycle progression in the etoposide-treated cells, we speculated that MPS1 inhibition enhanced centrosome abnormalities triggered by etoposide, thereby leading to defective cell-cycle progression and aberrant mitosis. To test this hypothesis, we carried out a microscopic analysis of cellular centrosomes with γ-tubulin staining (Figure 5a). When SCCVII cells were treated with either etoposide or IR, the formation of supernumerary centrosomes was stimulated (Figure 5a-5c). Consistent results were obtained in EMT6 cells (Figure 5d). Next, we examined whether MPS1 inhibition affected supernumerary centrosome formation stimulated by etoposide or IR. As shown in Figure 5b and 5c, NMS-P715 (0.5 μM), as well as AZ3146 (0.5 μM), increased the proportion of cells carrying more than two centrosomes after etoposide treatment in SCCVII cells. In contrast, neither MPS1 inhibitor affected centrosome status after IR. Similarly, although MPS1 inhibition promoted supernumerary centrosome formation by etoposide in EMT6 cells, this effect was not seen in IR-treated cells (Figure 5d). These results suggest that MPS1 inhibition exacerbates centrosome abnormalities in cells treated with etoposide but not with IR.
Effect of MPS1 inhibitors on MC after genotoxic treatments. Genotoxic stress has been shown to induce MC, a type of cell death associated with centrosome abnormalities and aberrant mitosis (20, 21). Since etoposide-induced, but not IR-induced, supernumerary centrosome formation was significantly enhanced by MPS1 inhibition, we examined the effect of the MPS1 inhibitors on the induction of MC after etoposide or IR treatment. After cellular nuclei staining, the cells with features of aberrant mitotic nuclei (micro-, multilobular and fragmented nuclei) were scored as cells undergoing MC. As shown in Figure 6a, when compared to control cells, etoposide, as well as IR, resulted in the induction of MC in SCCVII cells. Furthermore, NMS-P715 (0.5 μM), as well as AZ3146 (0.5 μM), significantly enhanced MC after etoposide treatment in SCCVII cells (Figure 6b and c). In contrast, neither MPS1 inhibitor affected MC after IR. Similarly, although MPS1 inhibition promoted the etoposide-induced MC in EMT6 cells, this effect was not seen in IR-treated cells (Figure 6d). These findings show that MPS1 inhibition enhances MC induced by etoposide, but not by IR, implying that MC induction is, at least in part, responsible for the differences in the sensitizing effects of the MPS1 inhibitors when treated with genotoxic agents.

Efficacy of MPS1 inhibitors to inactivate SAC. (a) Nocodazole-induced mitotic arrest in SCCVII cells. Representative flow cytometry profiles of p-H3 levels in the cells treated with vehicle (gray) or nocodazole (black) for 8 h. Dotted line represents the positive-negative threshold. (b and c) Nocodazole-arrested SCCVII cells were treated with NMS-P715 (b) or AZ3146 (c) at various concentrations for 2 h. Percentage of p-H3-positive cells was determined by flow cytometry. Data are expressed as mean±S.D. of three experiments. *p<0.05, **p<0.01, when compared to the MPS1 inhibitor (–) (Dunnett's test).
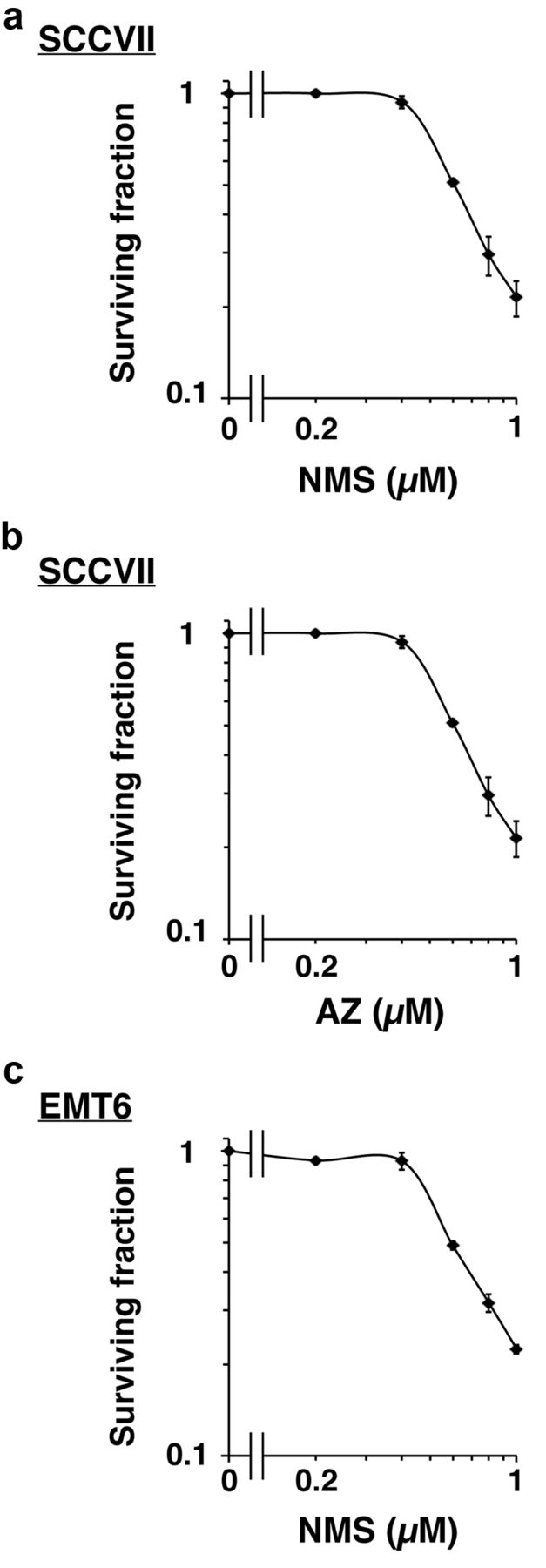
Cytotoxicity of MPS1 inhibitors. The cytotoxicity of MPS1 inhibitors was examined using a clonogenic survival assay. Cells were treated with NMS-P715 or AZ3146 at the indicated concentrations for 24 h and cultured for 6 days. Colonies containing more than 50 cells were scored as surviving colonies and clonogenic survival curves were obtained. (a) SCCVII cells±NMS-P715. (b) SCCVII cells±AZ3146. (c) EMT6 cells±NMS-P715. Data are expressed as mean±S.D. of three experiments.

Effect of MPS1 inhibitors on cellular sensitivity to genotoxic treatments. Cells were treated with either NMS-P715 (0.5 μM) or AZ3146 (0.5 μM) as follows. For etoposide treatment (left), cells were treated with etoposide in the presence or absence of the MPS1 inhibitors for 24 h. For IR (right), cells were irradiated, followed by incubation with the MPS1 inhibitors for 24 h. (a) SCCVII cells±NMS-P715. (b) SCCVII cells±AZ3146. (c) EMT6 cells±NMS-P715. Data are expressed as mean±S.D. of three experiments.

Effect of MPS1 inhibitors on cell-cycle distribution after genotoxic treatments. (a) Representative flow cytometry profiles showing the cell-cycle distribution of SCCVII cells treated with etoposide (400 nM) or IR (5 Gy). (b-d) Quantitative analysis of the effect of MPS1 inhibitors on cell-cycle distribution after genotoxic treatments (etoposide: 400 nM for SCCVII cells, 500 nM for EMT6 cells; IR: 5 Gy). (b) SCCVII cells±NMS-P715 (0.5 μM). (c) SCCVII cells±AZ3146 (0.5 μM). (d) EMT6 cells±NMS-P715 (0.5 μM). Data are expressed as mean±S.D. of three experiments.

Effect of MPS1 inhibitors on centrosome status after genotoxic treatments. (a) Representative images of the centrosomes in SCCVII cells treated with etoposide (400 nM) or IR (5 Gy). After treatment, centrosomes were visualized by γ-tubulin staining. The dashed circles represent nuclear outlines. Scale bar, 10 μm. (b-d) Quantitative analysis of the effect of MPS1 inhibitors on centrosome status after genotoxic treatments (etoposide: 400 nM for SCCVII cells, 500 nM for EMT6 cells; IR: 5 Gy). After γ-tubulin staining, a minimum of 100 cells was evaluated and the percentage of cells with more than two centrosomes was determined. (b) SCCVII cells±NMS-P715 (0.5 μM). (c) SCCVII cells±AZ3146 (0.5 μM). (d) EMT6 cells±NMS-P715 (0.5 μM). Data are expressed as mean±S.D. of three experiments. *p<0.05, **p<0.01, when compared to the MPS1 inhibitors (–) (Student's t-test). n.s., Not significant.
Discussion
In the present study, we revealed that MPS1 inhibition enhanced the etoposide-induced MC (Figure 6). MC is a form of cell death associated with aberrant mitosis due to uncoordinated or improper M phase progression and considered the major cell death mechanism after genotoxic stress, especially in apoptosis-impaired tumour cells (22-24). Therefore, the increase of MC by MPS1 inhibition is likely to be, at least in part, responsible for the potentiation of the etoposide-induced cytotoxicity observed in this study (Figure 3). It is known that supernumerary centrosome formation contributes to the induction of MC through abnormal chromosomal division or failure of cytokinesis (25, 26). Consistent with this finding, we found that MPS1 inhibition significantly increased supernumerary centrosome formation by etoposide (Figure 5). As the presence of supernumerary centrosomes is associated with aberrant mitosis via the formation of a multipolar spindle (20, 21), it seems plausible that MPS1 inhibition causes defective mitosis by enhancing the etoposide-triggered centrosome abnormality. The abnormality in centrosome numbers is mainly due to centrosome duplication system dysfunction or other defects due to irregular cell cycle progression (27). Similar to microtubule poisons, such as paclitaxel, etoposide has been shown to cause mitotic arrest via the activation of SAC (28). In cells undergoing SAC-dependent mitotic arrest, MPS1 inhibition is expected to inactivate SAC and allow cells to restart their cell cycle. In the present study, we observed that etoposide increased the cell population in the G2/M phase, while co-treatment with the MPS1 inhibitors further increased it (Figure 4). If the increased G2/M cell population by etoposide is due to SAC activation and its inactivation releases cells from G2/M arrest, our data appear to be contradictory to the assumption that MPS1 inhibition inactivates the etoposide-induced SAC. We speculate that co-treatment of the MPS1 inhibitor with etoposide promoted premature cell-cycle re-entry and induced aberrant mitosis via the formation of a multipolar spindle, resulting in the generation of irregular tetraploid (4N) cells but not normal G2/M cells. Collectively, this line of evidence suggests that MPS1 inhibition (i) prematurely inactivates SAC triggered by etoposide; (ii) causes aberrant cell-cycle progression and disturbs the centrosome duplication system, leading to supernumerary centrosome formation; (iii) results in defective mitosis and, consequently, the promotion of MC.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Effect of MPS1 inhibitors on MC after genotoxic treatments. (a) Representative images of cell nuclei in SCCVII cells treated with etoposide (400 nM) or IR (5 Gy). After treatments, cell nuclei were stained with DAPI. Scale bar, 10 μm. (b-d) Quantitative analysis of the effect of MPS1 inhibitors on MC after genotoxic treatments (etoposide: 400 nM for SCCVII cells, 500 nM for EMT6 cells; IR: 5 Gy). After DAPI staining, a minimum of 100 cells was evaluated and the percentage of cells undergoing MC was determined. (b) SCCVII cells±NMS-P715 (0.5 μM). (c) SCCVII cells±AZ3146 (0.5 μM). (d) EMT6 cells±NMS-P715 (0.5 μM). Data are expressed as mean±S.D. of three experiments. *p<0.05, **p<0.01, when compared to the MPS1 inhibitors (–) (Student's t-test). n.s., not significant.
In this study, we observed that IR (5 Gy) induced MC in about 80% of the irradiated cells (Figure 6). This was nearly equal to the level of the radiation-induced cell death evaluated by the clonogenic survival assay (Figure 3), indicating that MC accounts for the majority of radiation-induced cell death. However, etoposide caused approximately 40-50% of MC, whereas this same treatment resulted in 60-70% of cell death (Figures 3 and 6). Therefore, although MC appears to be the major route for cell death after etoposide treatment, other cell death pathways may also be involved in the etoposide-induced cell death. Etoposide has been shown to induce apoptosis and cellular senescence (20, 29). It remains elusive how MPS1 inhibition influences these MC-independent cell death pathways after etoposide treatment; therefore, further research is needed to decipher these pathways.
We found that MPS1 inhibition had a low impact on cellular radiosensitivity (Figure 3), although IR, like etoposide, is a well-known genotoxic agent. Contrary to our results, a recent study by Maachani et al. demonstrated that the combination of NMS-P715 with IR increases MC in the LN18 human glioblastoma cell line, indicating that MPS1 inhibition enhances cellular radiosensitivity (16). In the present study, we also observed that IR caused MC in SCCVII and EMT6 cells, which was, however, unaffected by MPS1 inhibition (Figure 6). This inconsistency may be partially explained by species-dependent differences in the stringency of SAC to maintain cells in mitotic arrest. Previously, Kung et al. found that there are significant differences between human and rodent cells in their ability to progress into a subsequent cell cycle in the presence of the spindle poison colcemid (30). They demonstrated that, when prolonged mitotic arrest occurs, rodent cells tend to skip mitosis and continue cell-cycle progression, whereas human cells remain arrested in the mitotic phase, suggesting that rodent cells are more prone to slip from SAC than human cells. Because SCCVII and EMT6 cells are derived from mouse tumours, it is possible that these murine cells skipped SAC triggered by IR, diminishing the effectiveness of the MPS1 inhibitors.
In conclusion, in this study, we demonstrated that MPS1 inhibition increased cellular sensitivity to etoposide, but not to IR, in the murine tumour cell lines, SCCVII and EMT6. In addition, MPS1 inhibition altered the cell-cycle progression and exacerbated centrosome abnormalities, resulting in an enhanced MC induction when cells are exposed to etoposide but not to IR. These results suggest that MPS1 inhibition has different effects depending on the type of genotoxic stress. Additionally, we demonstrated that MPS1 inhibition, in combination with etoposide, promotes aberrant mitosis and, consequently, induction of tumour cell death.
Acknowledgements
This work was supported, in part, by the Japanese Society for the Promotion of Science KAKENHI (Grant numbers 26461875 [TY], 25861045 [HY], and 24659551 [OI]), the Takeda Science Foundation [TY], and a research grant for the Study of the Health Effects of Radiation Organized by the Ministry of the Environment, Japan [OI and TY].
Footnotes
Conflicts of Interest
The Authors declare no conflict of interest.
- Received March 30, 2016.
- Revision received April 28, 2016.
- Accepted May 8, 2016.
- Copyright© 2016 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved