

Abstract
Background/Aim: The phosphatidylinositol-3-kinase (PI3K) signaling pathway is frequently activated in cancer. Buparlisib (BKM120), an oral pan-PI3K inhibitor, inhibits proliferation of human cancer in preclinical models. Studies of buparlisib in Western and Japanese adults with advanced solid tumors established a recommended dose of 100 mg/day and showed an acceptable safety profile and evidence of efficacy. This phase I dose-escalation/expansion study aimed to establish the maximum tolerated dose (MTD) of single-agent, once daily oral buparlisib in Chinese patients with advanced solid tumors. Materials and Methods: Patients (n=32; primary tumor site: lung (n=15), breast (n=10) or head and neck (n=7); ≥2 prior lines of antineoplastic therapy (n=26)) received 80 mg (n=15) or 100 mg (n=17) daily buparlisib. Results: Five patients experienced dose-limiting toxicities: grade (G)3 depression (n=1), G2 hyperglycemia (n=3) and G3 hyperglycemia (n=1). Most frequent buparlisib-related adverse events were hyperglycemia (n=18; 56%), alanine aminotransferase (ALT) and aspartate aminotransferase (AST) increase (n=9; 28%), as well as anxiety (n=6; 19%); most common buparlisib-related G3/4 adverse events: hyperglycemia (n=3; 9%), ALT and AST increase (n=2; 6%), as well as gamma-glutamyltransferase increase (n=2; 6%). Best response was stable disease (SD) in 10 patients (31%). Conclusion: The MTD of buparlisib was declared as 100 mg/day. Safety, efficacy and pharmacokinetic data from this study were similar to those previously reported in Western and Japanese populations.
- Buparlisib
- phosphatidylinositol-3-kinase
- breast cancer
- head and neck cancer
- lung cancer
The phosphatidylinositol-3-kinase (PI3K) signaling pathway regulates a diverse range of cellular processes, including proliferation, survival, protein synthesis, glucose metabolism, migration and angiogenesis (1). The PI3K pathway is the most frequently activated signaling pathway in human cancer. Increases in pathway activity can occur via several mechanisms, including mutation or amplification of upstream regulators, including growth factor receptors, activating mutations in the PIK3CA gene that encodes the PI3K p110α isoform and loss of the negative regulator of PI3K signaling and tumor suppressor phosphatase and tensin homolog (PTEN) (1-4). Activation of the PI3K pathway plays an important role in tumor formation and growth and can cause resistance to anticancer agents, including chemotherapy, endocrine therapy and other targeted therapies (5, 6). The critical signaling role played by PI3K in cancer cells makes this enzyme a valid therapeutic target.
Buparlisib (BKM120) is an oral, pan-PI3K inhibitor that targets all four class I isoforms of PI3K and can inhibit both wild-type and mutant versions of the protein at nanomolar concentrations (7). Buparlisib decreases PI3K signaling, promotes apoptosis and prevents cell proliferation in a variety of human cancer cell lines with and without constitutive PI3K activation and causes tumor growth inhibition and regression in xenograft tumor models (7). The first-in-human phase I study of buparlisib in Western adult patients with advanced solid tumors identified the maximum tolerated dose (MTD) as 100 mg/day during dose escalation (8), that was confirmed in the subsequent dose-expansion part of the study (9). A similar phase I study of buparlisib in adult Japanese patients with advanced solid tumors also established a recommended dose of 100 mg/day (10). Buparlisib showed an acceptable safety profile as a single agent in both studies with common treatment-related adverse events (AEs), including hyperglycemia, diarrhea, rash, decreased appetite, mood alteration and abnormal hepatic function (8-10). Single-agent buparlisib showed preliminary evidence of efficacy in both patient populations in metastatic breast cancer and other types of solid tumors (8-10). This phase I study was undertaken to establish a recommended dose for future development of buparlisib in the Chinese population.
Materials and Methods
Study design and treatment. This was a phase I, open-label, dose-escalation and -expansion study (ClinicalTrials.gov Identifier: NCT01626209) of single-agent buparlisib administered orally once daily in Chinese patients with advanced solid tumors. The primary objective was to establish the MTD/recommended phase II dose (RP2D) of single-agent buparlisib in adult Chinese patients with advanced solid tumors. Secondary objectives were to investigate the safety, tolerability, pharmacokinetic (PK) profile and clinical efficacy of buparlisib as a single agent in this patient population. During the dose-escalation period, at least three evaluable patients were to be enrolled at each dose level, to be expanded to -at least-six evaluable patients at the level in which the MTD/RP2D was reached. Two dose levels were to be tested: 80 mg/day and 100 mg/day, each in 28-day treatment cycles. An adaptive Bayesian logistic regression model for dose escalation with overdose control (EWOC) was used to guide the dose-escalation part of the study to estimate the MTD/RP2D (11, 12). The MTD was defined as the highest drug dosage not causing a medically unacceptable dose-limiting toxicity (DLT) in more than 33% of the treated patients in the first cycle of treatment and was estimated based on clinical and PK considerations. Patients were treated until disease progression, unacceptable toxicity, investigator's decision or until they refused further study treatment. Informed consent was obtained from each patient prior to enrollment and the study protocol and all amendments were reviewed by the Independent Ethics Committee or Institutional Review Board at each study site. The study was conducted according to the International Conference on Harmonization Harmonized Tripartite Guidelines for Good Clinical Practice, with applicable local regulations (including European Directive 2001/20/EC and US Code of Federal Regulations Title 21) and with the ethical principles laid down in the Declaration of Helsinki.
Patients' eligibility. Adult Chinese patients with advanced unresectable breast cancer or advanced carcinoma with squamous cell histology (including non-small cell lung cancer, squamous cell carcinoma of the head and neck and esophageal cancer) whose disease had progressed on (or who had been unable to tolerate) standard therapy or for whom no standard therapy existed were eligible for this study. Other key inclusion criteria were measurable and/or non-measurable disease as per Response Evaluation Criteria in Solid Tumors (RECIST) v1.1 guidelines; availability of a representative archival or fresh tumor biopsy; Eastern Cooperative Oncology Group (ECOG) performance status (PS) ≤2; and adequate bone marrow and organ function. Patients were excluded if they had received previous treatment with a PI3K inhibitor; symptomatic central nervous system (CNS) metastases; concurrent treatment with another approved or investigational antineoplastic therapy; increasing or chronic treatment with immunosuppressive agents, such as corticosteroids; active cardiac disease or a history of cardiac dysfunction; Fridericia's corrected QT >480 ms on screening electrocardiogram; impairment of gastrointestinal (GI) function or GI disease that may significantly alter the absorption of buparlisib; unresolved diarrhea of Common Terminology Criteria for Adverse Events (CTCAE) grade 2 or greater; patient health questionnaire-9 (PHQ-9) score ≥12 or a positive response to PHQ-9 question 9 relating to suicidal ideation; generalized anxiety disorder 7 (GAD-7) mood scale score ≥15; a documented or current major depressive episode or other specified psychiatric disorders.
Definition of dose-limiting toxicity (DLT). A DLT was defined as an AE or abnormal laboratory value that fulfilled all of the following criteria: unrelated to disease, disease progression or concomitant medications; occurrence within 28 days following the first dose of buparlisib; meeting any of the criteria in Table I.
Study assessments. Safety assessments consisted of monitoring by physical examination vital signs, weight, ECOG PS, electrocardiogram, laboratory evaluations and assessment of patient-rated mood scales (PHQ-9 and GAD-7), in addition to the recording of all serious and non-serious AEs using CTCAE version 4.03. Laboratory evaluations included hematology, blood chemistry, urinalysis and glucose monitoring. For PK assessments, blood samples for buparlisib plasma concentration–time profiles were collected for all study patients on Days 1, 8 and 28 of Cycle 1 pre-dose and at 0.5, 1, 1.5, 2, 3, 4, 6, 8 and 24 hours post-dose, as well as on Day 1 of Cycle 3 and every other cycle thereafter, pre-dose and 2-4 h post-dose. Buparlisib in plasma was analyzed by liquid chromatography-tandem mass spectrometry (limit of quantification 1.0 ng/ml). PK parameters were determined using non-compartmental methods (WinNonlin® Pro version 5.2 or Phoenix® WinNonlin® version 6.0 (Pharsight, Mountain View, CA, USA)). For biomarker assessments, archival or fresh tumor samples were investigated for aberrations of the PI3K pathway by analysis of PIK3CA mutations using gene sequencing and PTEN protein expression level using immunohistochemistry. For efficacy assessments, radiologic assessment of tumor lesions using computed tomography (CT) or magnetic resonance imaging (MRI) was performed at baseline and every 8 weeks thereafter. Tumor response was assessed using RECIST v1.1.
Results
Patients' characteristics and treatment. Between July 2012 and April 2014, 32 Chinese patients with advanced solid tumors were enrolled and treated with buparlisib at three centers in China: 15 patients received 80 mg buparlisib and 17 patients received 100 mg buparlisib once daily (Table II). The median age of patients was 52 (range=24-75) years, 56% (18 of 32) were female and 91% (29 of 32) had an ECOG PS of 0-1. The predominance of tumor type was non-small cell lung cancer (n=15), breast (n=10) or squamous cell cancer of head and neck (n=7). This patient population was heavily pre-treated, with 81% (n=26) and 38% (n=12) having received at least two or four prior lines of antineoplastic therapy, respectively, including at least one line of therapy in the metastatic setting in 84% (n=27) of patients. Seven patients in the study were reported to have PI3K pathway activation: two due to PIK3CA mutation, four due to loss of PTEN expression and one due to both a PIK3CA mutation and loss of PTEN expression. Nineteen patients (59%) had progressed within 12 months following the initial diagnosis.
Criteria for defining dose-limiting toxicities.
Baseline patients' and disease characteristics.
DLTs and MTD. Twelve patients were included in the dose-determining set (DDS) used to establish the MTD: four in the 80-mg buparlisib group and eight in the 100-mg buparlisib group. Five patients experienced DLTs during the study. Two DLTs occurred in the dose-escalation part of the study and were grade 3 depression (n=1, 80-mg group) and grade 2 hyperglycemia (n=1, 100-mg group). Based on the results from patients in the DDS, the MTD of buparlisib was declared as 100 mg/day. Additional patients were enrolled into the dose-expansion part of the study in parallel dose groups (80 mg and 100 mg) for evaluation of the safety and tolerability of buparlisib. Three DLTs occurred in the dose-expansion part of the study; all were hyperglycemia in the 80-mg group (n=2 grade 2 hyperglycemia; n=1 grade 3 hyperglycemia). The MTD of buparlisib was confirmed at 100 mg/day with an overall frequency of DLTs at this dose level of 3%.
Safety and tolerability. All patients experienced at least one AE regardless of relationship to the study drug. These included hyperglycemia in 19 patients (59%), increased alanine aminotransferase (ALT) in 14 patients (44%), increased aspartate aminotransferase (AST) in 13 patients (41%; 12 patients had an increase in both ALT and AST (38%)) and decreased appetite in 10 patients (31%). Study drug-related AEs were seen in 28 patients (88%), including hyperglycemia in 18 patients (56%), ALT and AST increase in the same nine patients (28% each), as well as anxiety in six patients (19%; Table III).
Eight patients (25%) experienced grade 3/4 AEs that were suspected to be related to buparlisib; these included hyperglycemia in three patients (9%), ALT and AST increase in the same two patients (6% each) and gamma-glutamyltransferase increase in two patients (3%). The frequency of treatment-related grade 3/4 AEs was similar at the two dose levels tested: 27% of patients treated at 80 mg and 24% treated at 100 mg experienced grade 3/4 AEs. Four patients in each dose group experienced at least one serious AE (SAE) regardless of relationship to the study drug. Two patients (12%) in the 100-mg group experienced SAEs suspected to be study drug-related, including one patient with a grade 3 suicide attempt and one patient with grade 2 pneumonitis.
Irrespective of causality, AEs led to buparlisib discontinuation in seven patients (22%). These included three patients in the 80-mg group (one case each of grade 3 pneumonia, grade 4 dyspnea and grade 4 hemoptysis) and four patients in the 100-mg group (one case each of grade 2 pulmonary tuberculosis, grade 2 hyperglycemia, grade 2 pneumonitis and grade 3 suicide attempt). In addition, 11 patients (34%) experienced AEs requiring dose interruption or dose adjustments: five patients (33%) in the 80-mg group and six patients (35%) in the 100-mg group; of which hyperglycemia in four patients was the most frequent (three patients in the 80-mg group and one patient in the 100-mg group). Other reasons for dose interruption or change were pyrexia, depression, anemia, AST or ALT increase, decreased platelets, thrombocytopenia, hiccups, decreased appetite and dyspnea (n=1 each). Eight deaths (25%; four in each dose group) occurred during the study; none were suspected to be study drug-related. Seven patients died due to disease progression and one patient died due to pneumonia. The median duration of exposure to buparlisib was 48 days in the 80-mg group and 57 days in the 100-mg group. The median relative dose intensity for all patients was 100%.
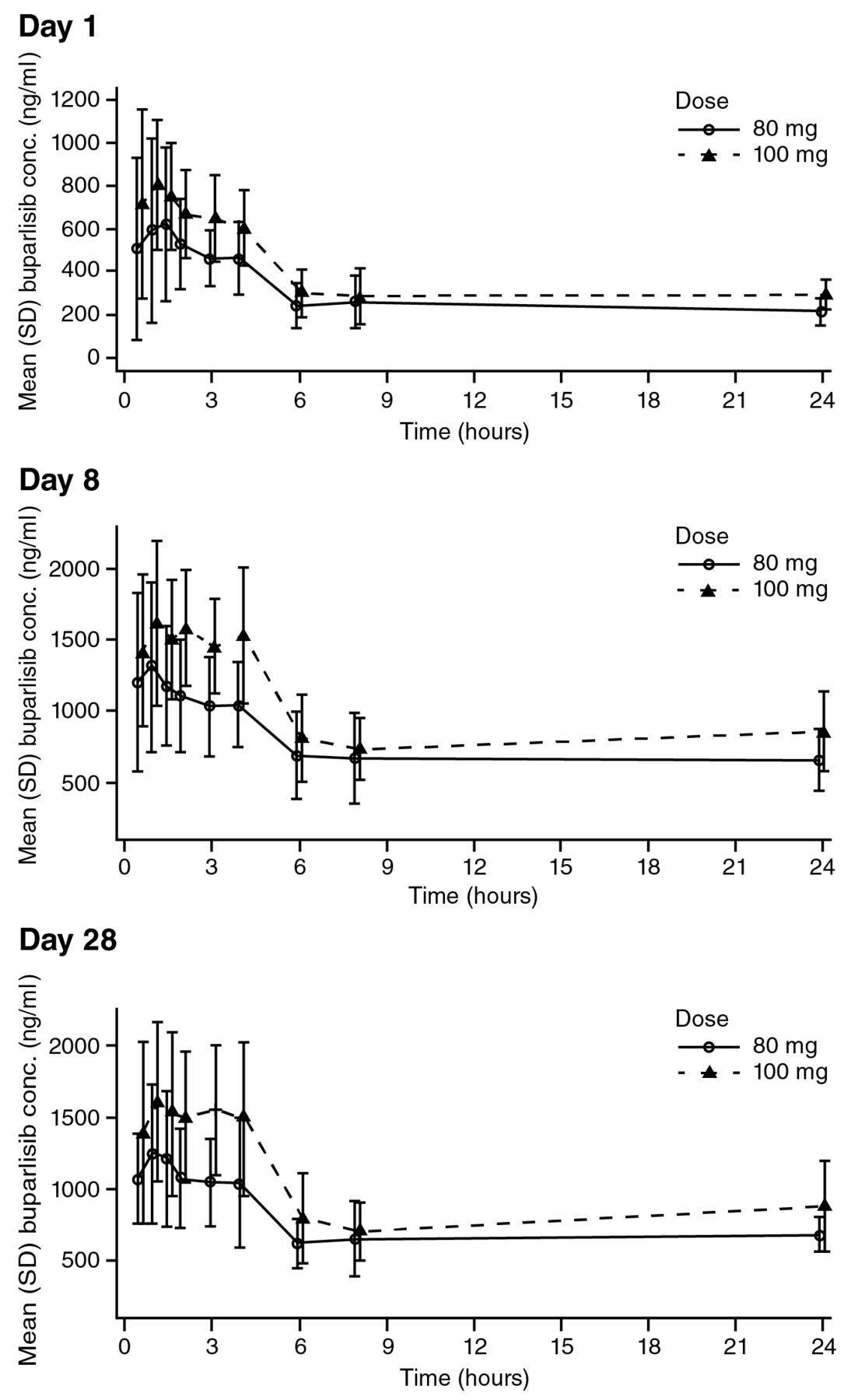
Pharmacokinetics. After both single and repeated doses, the absorption of buparlisib was rapid in both treatment groups with a median Tmax of 1-1.7 h (Table IV). Following a single dose of 80 mg buparlisib, the mean Cmax was 838 ng/ml, which is approximately 80% of the mean Cmax observed after a single 100-mg dose, suggesting a proportional increase of Cmax with dose (Table IV). After repeated daily doses of buparlisib, mean Cmax was similar at Day 8 and Day 28 for each of the two dose levels with higher values for the 100-mg dose (Table IV). Similarly, the area under the curve (AUC)0-24 values were similar at Day 8 and 28 for each of the two doses, again with higher values in the 100-mg dose (Table IV). These results are consistent with a Cmax increase proportional to dose and with steady-state for buparlisib being reached after seven days of continuous dosing. The accumulation ratio, calculated based on the AUC0-24 ratio after repeated and single doses, was approximately 2.8 for both doses. Buparlisib displayed linear pharmacokinetics with a similar clearance at the 80-mg and 100-mg doses. The effective buparlisib half-life was also similar across doses, being between 36 and 39 h. The mean buparlisib plasma concentration profiles on Cycle 1 Days 1, 8 and 28 for each dose group are shown in Figure 1.
Most common adverse events (>10%) thought to be related to study treatment.
Pharmacokinetic profile of oral buparlisib in Chinese patients in Cycle 1.
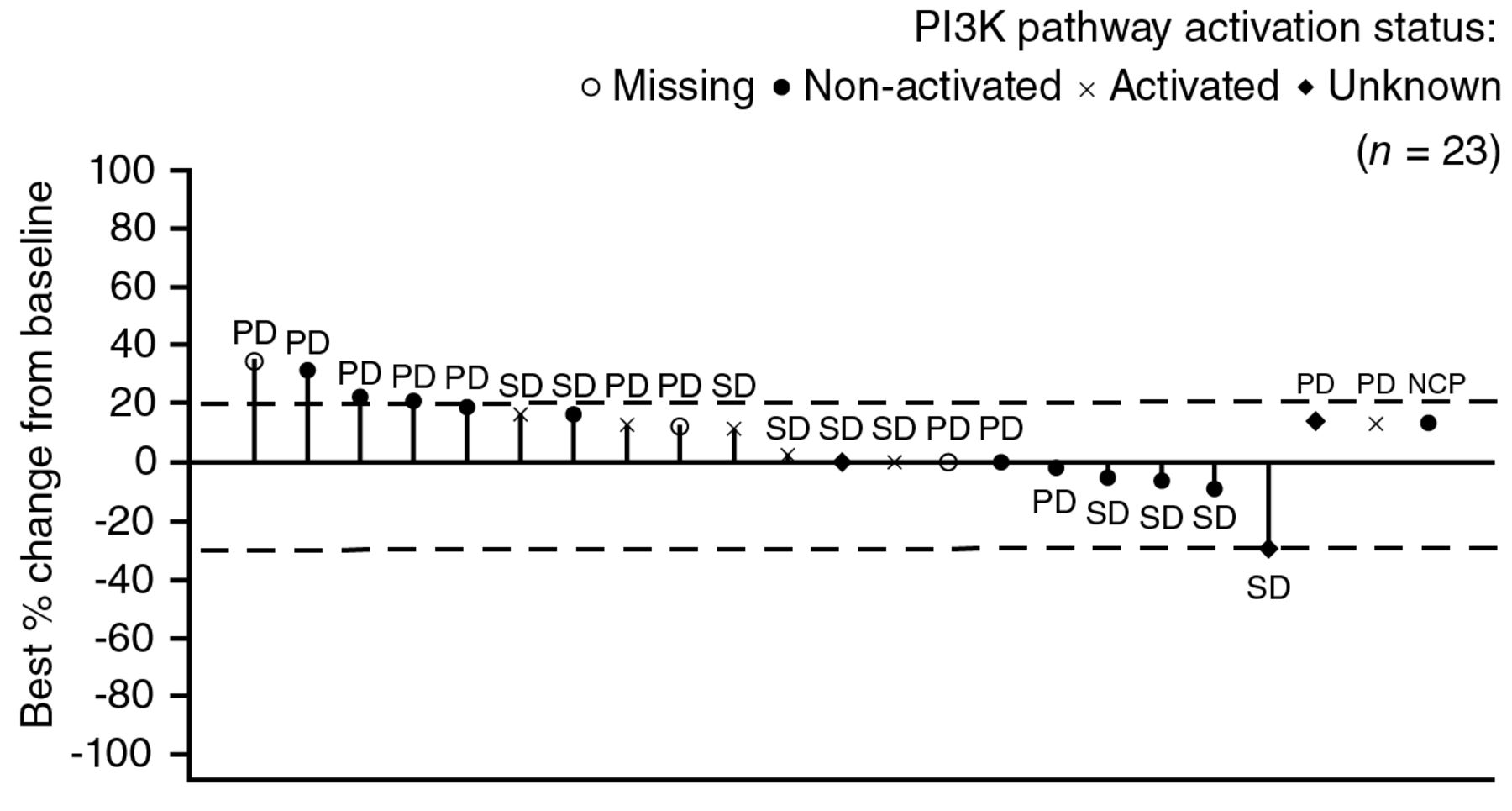
Efficacy. The reported antitumor activity of single-agent buparlisib was marginal. No patient achieved a complete response (CR) or partial response (PR) to buparlisib during the study (Table V). However, 10 patients (31%) exhibited stable disease (SD) as their best response (two patients (13%) in the 80-mg group and eight patients (47%) in the 100-mg group) and one patient (6%) with non-measurable disease (in the 100-mg group) had non-CR/non-PD. Nine patients (28%) were non-evaluable for best overall response due to the absence of a valid post-baseline assessment. Overall, the disease control rate (SD + non-CR/non-PD) was 34%: 13% in the 80-mg group and 53% in the 100-mg group. The best percentage changes from baseline by PI3K pathway activation status and dose level are presented in Figure 2. The median time to disease progression was 1.84 months (95% confidence interval (CI)=0.92-3.38) for the 80-mg group and 2.96 months (95% CI=1.71-3.61) for the 100-mg group.
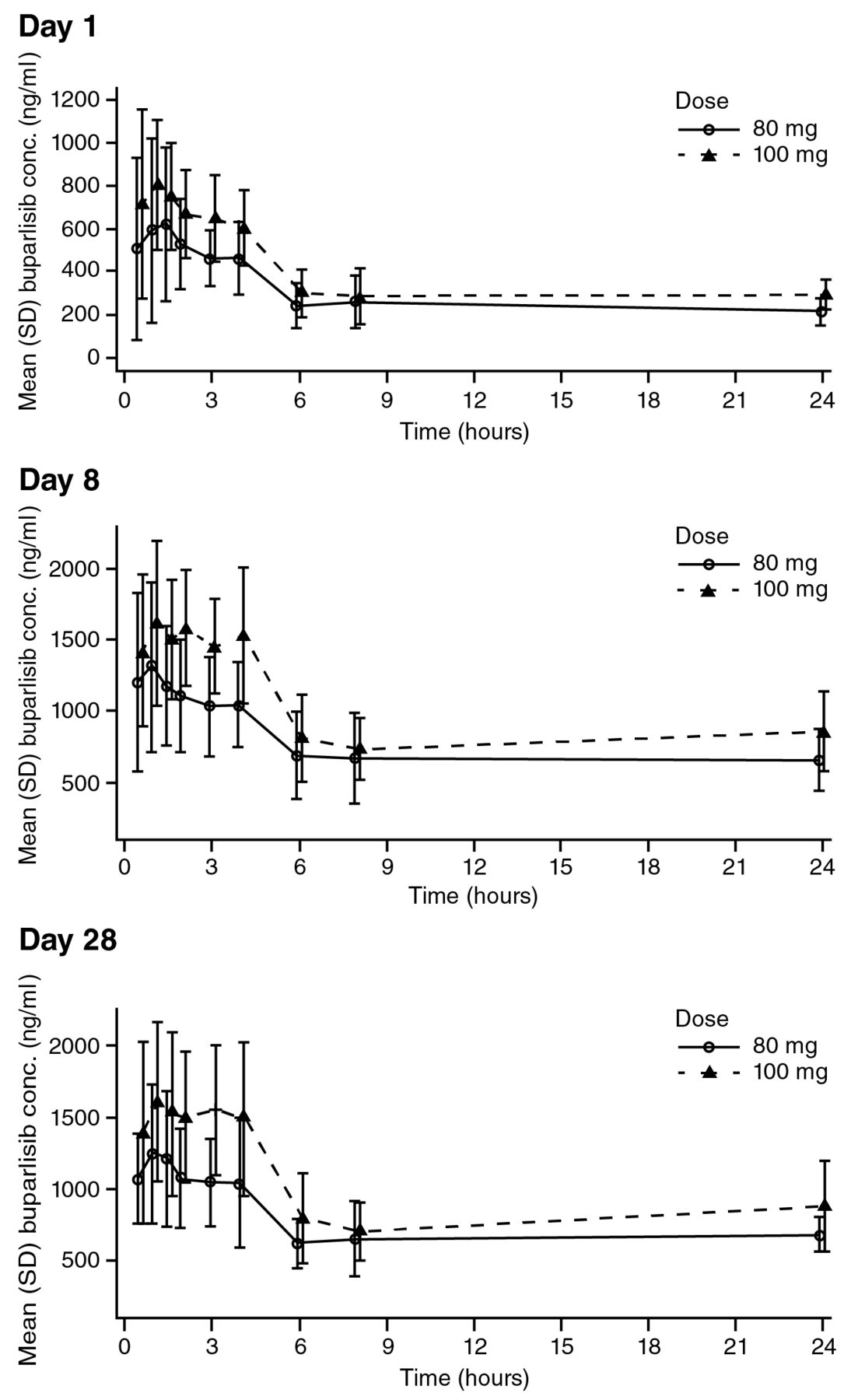
Linear plasma concentration profiles for buparlisib in Chinese patients in Cycle 1. SD, Standard deviation.
{kind=link}
{kind=link}
Radiologic response to buparlisib with corresponding status of tumor PI3K activation status. CR, Complete response; NCP, non-CR/non-PD; PD, progressive disease; PI3K, phosphatidylinositol-3-kinase; PR, partial response, SD, stable disease. Patients with missing best percentage change from baseline and unknown overall response are not included. Unknown indicates patients not qualifying for confirmed CR or PR and without SD after more than 6 weeks or early progression within the first 12 weeks. Missing line denotes a missing best percentage change from baseline.
Discussion
The primary objective of this phase I study of single-agent buparlisib in adult Chinese patients with advanced solid tumors was to establish the MTD and/or RP2D for this agent. Starting at 80 mg/day (a dose selected based on data for single-agent buparlisib in phase I studies in Western and Japanese patient populations) (8, 10), dose escalation established an MTD/RP2D for buparlisib in Chinese patients as 100 mg/day. This is the same dose as that established for single-agent buparlisib in Western and Japanese patients (8, 10). The posterior distribution of DLT rates indicated that 100 mg/day satisfied the EWOC criteria for the recommended dose and, together with safety and PK data, was used to determine the MTD.
The PK profiles observed in the Chinese patients treated with 100 mg/day buparlisib in this study are similar to those observed in Western and Japanese patients (8, 10). Buparlisib has been found to be rapidly absorbed after single and repeated doses, with a Tmax of 1-3 h, in all three studies. The effective half-life of buparlisib was approximately 40 h in Western and Japanese patients compared with 36-39 h in Chinese patients; steady-state for buparlisib is reached after seven days of continuous dosing in all patient populations studied to date (8, 10). After 28 days of buparlisib treatment, mean Cmax and AUC0-24 were similar across Western (Cmax 1850 ng/ml; AUC0-24 22,500 ng•h/ml), Japanese (Cmax 1790 ng/ml; AUC0-24 25,000 ng•h/ml) and Chinese patients (Cmax 1880 ng/ml; AUC0-24 22,400 ng•h/ml) (8, 10).
Clinical responses to buparlisib in Chinese patients with advanced solid tumors.
Safety results from this study were also similar to those recorded in Western and Japanese populations (8-10). Three main categories of AEs suspected to be related to buparlisib were: hyperglycemia, liver function abnormalities and mood disorders. Hyperglycemia is considered an on-target, class effect of PI3K inhibitors due to disrupted insulin signaling and reduced glucose uptake and glycogen synthesis as a result of PI3K inhibition (4, 13, 14). Other PI3K pathway inhibitors for which hyperglycemia has been reported include PKI-587 (PF-05212384) and BEZ235 (15, 16). In this study, hyperglycemia was the most frequently reported AE suspected to be related to treatment, reported in 18 patients (56%; grade 3/4 in three patients, 9%; Table III) and was the most frequent reason for dose interruption. This was slightly higher than the frequency of hyperglycemia suspected to be related to treatment reported in the first-in-human phase I study of buparlisib in Western patients, which was 31% overall (9). Only one case of grade 3/4 hyperglycemia (7%; irrespective of relationship to study drug) was reported in the phase I study of buparlisib in Japanese patients but the number of patients in that study was small (n=15) (10). Hyperglycemia was largely manageable with oral antidiabetic agents, such as metformin, and/or with insulin and dose interruptions, combined with careful monitoring of blood glucose levels, as recommended following previous experience (8, 14).
Liver function abnormalities suspected to be related to buparlisib treatment were also frequent in this study: increased ALT and AST were seen in the same 9 patients (28%; grade 3/4 in the same two patients, 6%). Increases in transaminase levels have been observed in previous studies of buparlisib (40% grade 3/4 transaminase increases were reported in Japanese patients and grade 3/4 transaminase increases were also reported in Western patients) and other PI3K pathway inhibitors at varying frequencies (8, 10, 15, 17-22). Further studies may allow characterization of hepatic enzyme perturbation with buparlisib, which may be an on-target effect of inhibiting PI3K or some other biochemical effect of the compound on hepatocytes. An exploratory analysis into a possible relationship between PK parameters for buparlisib and any increase in risk for liver function test alteration is ongoing (Novartis, unpublished data).
Mood alterations, in the form of anxiety and depression, were recorded in this study. Mood disorders following buparlisib treatment have been observed in other early clinical studies of the agent (8-10) and other buparlisib studies have restricted the enrolment of patients deemed to be at increased risk of psychiatric AEs. Studies of buparlisib have also used the PHQ-9 (depression) and GAD-7 (anxiety) questionnaires to monitor patients and allow timely intervention. Under-activation of the PI3K/AKT/mammalian target of rapamycin (mTOR) pathway has been linked to anxiety and depression (23-25). Notably, decreases in the activity of PI3K and AKT and increases in PTEN protein levels were observed in the ventral prefrontal cortex of depressed suicide victims and depressed non-suicide subjects (26). Buparlisib has been shown to cross the blood–brain barrier and has demonstrated preliminary clinical activity in brain lesions of patients with breast cancer and non-small cell lung cancer, suggesting buparlisib-mediated inhibition of PI3K in the CNS (27). Mood disorders are also commonly associated with cancer diagnoses, with studies estimating that 30%-50% of cancer patients experience psychological symptoms, including adjustment disorders, anxiety or depression (28, 29). In this study, the incidence of depression and anxiety was 13% and 19%, respectively (Table III).
Preliminary signs of clinical efficacy were observed in this study, with 10 patients (31%) exhibiting stable disease. The disease control rate reported here (34%) is similar to rates seen with buparlisib in other patient populations: 41% and 40% in the Western and Japanese patient populations, respectively (9, 10). Although it is possible that buparlisib may show greater efficacy in patients with activated PI3K signaling, this study contained too few patients to determine any correlation between mutation status and response, as has been possible with other targeted therapies, such as the epidermal growth factor receptor (EGFR) inhibitor gefitinib, found to be relatively more effective in patients with EGFR-mutated lung cancer than EGFR-wild-type disease (30), with clear implications for patient selection. Results from previous trials of buparlisib and other PI3K inhibitors have also been inconclusive regarding a correlation between PI3K activation status and clinical response (8, 9, 19, 31-35). Notably, PI3K activation status in this and other clinical studies of PI3K pathway inhibitors has been assessed using PIK3CA mutation and/or loss of PTEN expression in predominantly archival tissue (8, 9, 19, 31, 32); however, it is known that PI3K activation can occur independently of these molecular alterations (1, 3) and that the mutational status of PI3K can change upon disease recurrence (36). Therefore, further research is needed to determine if PI3K activation status is a predictor of buparlisib response and whether selecting for patients with PI3K activation could improve outcomes with buparlisib. Moreover, additional efforts are required to identify patients with PIK3CA-mutant tumors just prior to the start of study treatment with PI3K inhibitors to provide a more accurate assessment of the potential benefit of these agents according to the current molecular status of the tumor. The development of combination strategies targeting multiple pathways is also a potential future approach to achieving more meaningful therapeutic effects given that the single-agent activity seen so far with PI3K inhibitors has been limited (37). In this regard, multiple clinical trials are underway, including phase III studies of buparlisib in combination with fulvestrant in patients with advanced or metastatic hormone receptor-positive breast cancer.
Conclusion
This phase I dose-escalation and -expansion study has established the MTD of buparlisib in adult Chinese patients with advanced solid tumors as 100 mg/day. Single-agent buparlisib, administered orally, once daily, had an acceptable safety profile and was generally well tolerated in this patient population. The PK profiles and the preliminary signs of clinical efficacy observed in this study of Chinese patients are similar to results from previous studies of buparlisib in Western and Japanese patient populations (8, 10).
Acknowledgements
The Authors would like to thank the patients who took part in the study and their families, as well as the staff who assisted with the study at each site. This study was sponsored by Novartis Pharmaceuticals Corporation that also provided financial support for medical editorial assistance. We thank Karen Beckett PhD and Abbie Saunders PhD for medical editorial assistance with this manuscript.
Footnotes
Compliance with Ethical Standards Disclosure of Potential Conflicts of Interest
L.T., T.D., V.D. and K.H. are full-time employees of Novartis Pharmaceuticals Corporation, while V.D. owns shares in Novartis Pharmaceuticals Corporation. Y.W., L.Z. and B.X. have no financial relationships to declare.
Ethical Approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed Consent
Informed consent was obtained from all individual participants included in the study.
- Received August 2, 2016.
- Revision received September 28, 2016.
- Accepted September 29, 2016.
- Copyright© 2016 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved