

Abstract
Mutation of Kirsten rat sarcoma viral oncogene homolog (KRAS) and chronic pancreatitis are the most common pathogenic events involved in human pancreatic carcinogenesis. In the process of long-standing chronic inflammation, aberrant metabolites of arachidonic acid play a crucial role in promoting carcinogenesis, in which the soluble epoxide hydrolase (sEH), as a pro-inflammatory enzyme, generally inactivates anti-inflammatory epoxyeicosatrienoic acids (EETs). Herein, we determined the effect of our newly-synthesized novel compound trans-4-{4-[3-(4-chloro-3-trifluoromethyl-phenyl)-ureido]-cyclohexyloxy}-pyridine-2-carboxylic acid methylamide (t-CUPM), a dual inhibitor of sEH and RAF1 proto-oncogene serine/threonine kinase (c-RAF), on inhibiting the development of pancreatitis and pancreatic intraepithelial neoplasia (mPanIN) in LSL-KrasG12D/Pdx1-Cre mice. The results showed that t-CUPM significantly reduced the severity of chronic pancreatitis, as measured by the extent of acini loss, inflammatory cell infiltration and stromal fibrosis. The progression of low-grade mPanIN I to high-grade mPanIN II/III was significantly suppressed. Inhibition of mutant Kras-transmitted phosphorylation of mitogen-activated protein kinase's kinase/extracellular signal-regulated kinases was demonstrated in pancreatic tissues by western blots. Quantitative real-time polymerase chain reaction analysis revealed that t-CUPM treatment significantly reduced the levels of inflammatory cytokines including tumor necrosis facor-α, monocyte chemoattractant protein-1, as well as vascular adhesion molecule-1, and the levels of Sonic hedgehog and Gli transcription factor (Hedgehog pathway). Analysis of the eicosanoid profile revealed a significant increase of the EETs/dihydroxyeicosatrienoic acids ratio, which further confirmed sEH inhibition by t-CUPM. These results indicate that simultaneous inhibition of sEH and c-RAF by t-CUPM is important in preventing chronic pancreatitis and carcinogenesis.
- pancreatitis
- carcinogenesis
- chemoprevention
- soluble epoxide hydrolase
- c-RAF
Pancreatic cancer is one of the most lethal malignant neoplasms, and chronic pancreatitis and mutant Kirsten rat sarcoma viral oncogene homolog (KRAS) gene are the most common events involved in pancreatic carcinogenesis, with more than 90% of human pancreatic carcinomas carrying the KRAS gene mutation (1, 2). Mutations of KRAS lead to constitutive activation of KRAS and persistent stimulation of downstream signaling pathways that initiate carcinogenesis via sustained proliferation, metabolic reprogramming, anti-apoptosis, remodeling of the tumor microenvironment, evasion of the immune response, cell migration and metastasis (3). The mutant RAS-activated RAF1 proto-oncogene serine/threonine kinase (c-RAF)-mitogen-activated protein kinase's kinase (MEK)-extracellular signal-regulated kinases (ERK) pathway appears crucial for initiating carcinogenesis, and using mutant Kras-driven tumorigenesis in mice, it has further been demonstrated that signaling through c-RAF-MEK-ERK, but not b-RAF, are essential for tumor initiation by mutant Kras, and c-RAF is responsible for transmitting signals from mutant KRAS to MEK-ERK (4). Therefore, development of c-RAF inhibitor would be crucial for suppressing mutant Kras-initiated carcinogenesis.
Long-standing chronic inflammation, including chronic pancreatitis, is a well-recognized risk factor for pancreatic cancer (1). Aberrant metabolites of arachidonic acid in the process of inflammation play a crucial role in promoting carcinogenesis (5). Three metabolic pathways are involved in arachidonic acid metabolism: cyclooxygenases (COX), lipoxygenases (LOX), and cytochromes P450 (CYP); non-steroidal anti-inflammatory drugs such as COX inhibitor are the most promising agents in cancer prevention (6, 7). However, the frequently severe side-effects of these agents, including gastrointestinal ulcer, potentially life-threatening bleeding and cardiovascular risks, often prohibit their widespread clinical use (6, 7). Thus, the development of an efficient anti-inflammatory agent with minimal side-effects is imperative. Epoxyeicosatrienoic acids (EETs) are CYP450-mediated epoxygenated products of arachidonic acid that have been demonstrated to have an efficient anti-inflammatory effect through reducing cytokine-induced endothelial cell adhesion molecule (VCAM) and reducing nuclear factor kappa-B kinase and nuclear factor kappa-B kinase inhibitor activity (8). Under physiological conditions, EETs are quickly inactivated by soluble epoxide hydrolase (sEH) that catalyzes their conversion into dihydroxyeicosatrienoic acids (DHETs) (9). sEH inhibitor results in stabilizing EETs and increasing the EET/DHET ratio. EETs have shown potent anti-inflammatory activity in various rodent inflammatory disease models, mainly via reducing the production of nitric oxide, pro-inflammatory lipid mediators, as well as inflammatory cell infiltration (8, 10, 11).
Recently we synthesized a unique compound called trans-5-{4-[3-(4-chloro-3-trifluoromethyl-phenyl)-ureido]-cyclohexyloxy}-pyridine-2 carboxylic acid methylamide (t-CUPM) via modifying the central phenyl ring of sorafenib (12). t-CUPM showed high potent activity against c-RAF and sEH (12).
In the present study, using a unique mutant Kras-initiated and caerulein-induced model of pancreatitis-carcinogenesis in LSL-KrasG12D/Pdx1-Cre mice, the effect of t-CUPM against the development of pancreatitis and murine pancreatic intraepithelial neoplasia (mPanIN) was examined by histopathological and immunochemical analyses. The plasma eicosanoid profile was analyzed using liquid chromatography/tandem mass spectrometry (LC-MS/MS).
Materials and Methods
Animal experiment. Pdx1-Cre mice were kindly provided by Dr. Lowy (University of Cincinnati). LSL-K-rasG12D mice were obtained from Mouse Repository, National Cancer Institute (Frederick, MD, USA) and kindly provided by Dr. T Jacks (Massachusetts Institute of Technology). All Pdx1-Cre and LSL-KrasG12D mice were bred and genotyped in our laboratory following the protocols provided by investigators (2). Mice were housed under pathogen-free conditions and with free access to water and food. All studies were conducted in compliance with the Northwestern University Institutional Animal Care and Use Committee guidelines (approved animal study protocol#: ASP# 2013-3085).
Four-week-old (n=20) LSL-KrasG12D/Pdx1-Cre transgenic mice, and wild-type mice (n=10) were treated with a single intraperitoneal injection of caerulein (250 μg/kg body weight, Sigma, St. Louis, MO, USA) to induce chronic pancreatitis, according to the published method with a slight modification (13, 14). t-CUPM was synthesized in Dr. Hammock's laboratory and provided for this study. Four weeks after caerulein induction, t-CUPM was administered to 10 LSL-KrasG12D/Pdx1-Cre transgenic mice and five wild-type mice in drinking water at a concentration of 8 mg l−1 (average body weight for 8-week-old mice is 25 g; fluid consumption is 3 ml, therefore the dose was 0.008 mg/ml ×3 ml/0.025 kg=1.0 mg/kg body weight). t-CUPM was dissolved in polyethylene glycol 400 (PEG400) and then diluted with water, and the final concentration of PEG400 in drinking fluid was 2.5% (v/v). 2.5% PEG400 was given to the remaining mice in drinking fluid as a vehicle control. All mice were treated for 4 weeks. AIN93M diet (Research Diet, New Brunswick, NJ, USA) was administered during the course of the experiment. The drinking fluids were changed every day and diet was replenished every 3 days. Food and water consumption were monitored every day and body weight was measured weekly.
Tissue preparation and histological analysis of chronic pancreatitis and mPanIN lesions. The pancreas and other key organs (including liver and spleen) were collected and weighed immediately after sacrificing the mice. Half of each organ was fixed in buffered formalin and routinely processed for paraffin sectioning. Serial paraffin sections (5 μm) were used for histopathological analysis, histochemical staining (Masson Trichrome) and immunohistochemical staining. Plasma and the other half of each organ were freshly collected and used for biochemical assays.
Chronic pancreatitis was blindly analyzed and graded using a semi-quantitative scoring system for the extent or areas of acini loss, number of inflammatory cells per high-power field (×40 objective lens) and the area and intensity of stromal fibrosis according to our established criteria (14). The chronic pancreatitis index (CPI) was expressed as a sum of scores for acini loss, inflammatory cell infiltration and stromal fibrosis. mPanIN (graded as mPanIN1, mPanIN2, and mPanIN3) and carcinoma were analyzed based on the consensus criteria for diagnosis and grading of mPanIN (15).
Immunohistochemistry. Immunohistochemical staining was performed according to our routine protocol using an avidin-biotin-peroxidase method (16). The primary antibodies included rabbit polyclonal anti-myeloperoxidase (MPO) (Abcam, Cambridge, MA, USA.), rat monoclonal antibody to murine MAC3 (Novus Biologicals, Littleton, CO, USA), mouse monoclonal anti-proliferating cell nuclear antigen (PCNA) (Calbiochem, Gibbstown, NJ, USA), goat polyclonal anti-cytokeratin 19 (Santa Cruz Biotechnology, Santa Cruz, CA, USA), mouse monoclonal anti-amylase (Santa Cruz Biotechnology), and rabbit monoclonal anti-phospho-ERK1/2 (Cell Signaling Technology, Boston, MA, USA) antibody. The appropriate biotinylated secondary antibody and avidin-biotin-peroxidase complex (Vector Lab, Burlingame, CA, USA) were used for detecting antigen–primary antibody complex. A characteristic brown color was developed by incubation with 3,3-diaminobenzidine substrate chromogen system (Sigma-Aldrich, St. Louis, MO, USA). The negative control was established by replacement of primary antibody with normal serum, and an appropriate positive control was used for each primary antibody. Specific antibody-labeled signals were analyzed under a Nikon research microscope (Nikon instrument Inc., Melville, NY, USA) for positive cell number and staining intensity.
Quantitative real-time polymerase chain reaction (PCR) assay. Total RNA was extracted from pancreatic tissue using the RNeasy® mini kit (Qiagen, Inc., Valencia, CA, USA) and the concentration was determined using a SmartSpec Plus Spectrophotometer (BioRad, Hercules, CA, USA). First-strand cDNA was synthesized using 1 μg of total RNA in a 20 μl reverse transcriptase reaction mixture using iScript™ cDNA synthesis kit (BioRad) according to the manufacturer's instructions. The region of tumor necrosis factor-α (Tnfa), interleukin-6 (Il6), monocyte chemoattractant protein-1 (Mcp1), vascular adhesion molecule 1 (Vcam1), sonic hedgehog (Shh) and Gli transcription factor (Gli) mRNA were amplified using a real-time PCR approach according to our previous published protocol (17, 18). All real-time PCR reactions and quantitations were performed using the MiniOpticon Real-Time PCR System (BioRad). Data of each mRNA expression are shown as the relative fold change normalized by that of glyceraldehyde 3-phosphate dehydrogenase (Gapdh).
Analysis of metabolic profile of oxylipin using LC-MS/MS. Serum specimens were spiked with 10 μl of 50 nM internal standard (d11-14, 15-dihydroxyeicosatrienoic acid (DHET), d11-11(12)-EET, d4-prostaglandin E2 (PGE2), d4-leukotriene B4 (LTB4), and d8-5-hydroxyeicosatetraenoic acid (HETE)) and were extracted by solid-phase extraction using Oasis HLB cartridges (3cc 60 mg; Waters, Milford, MA, USA). The HLB cartridges were first washed with 2 ml ethyl acetate, 2 ml methanol twice, and 2 ml of 95:5 v/v water/methanol with 0.1% acetic acid. Then, 200 μl serum samples were loaded onto the cartridges, which were then washed with 6 ml 95:5 v/v water/methanol with 0.1% acetic acid, and dried for 20 min with low vacuum. The target analytes were eluted with 0.5 ml methanol, followed by 2 ml of ethyl acetate into the tubes with 6 μl of 30% glycerol in methanol as the trap solution. The volatile solvents were evaporated using vacuum centrifugation (Speed-Vac, VWR, Chicago, IL, USA) until ~2 μl trap solution remained in the tube. The residues were dissolved in 50 μl of methanol containing 200 nM internal standards II (1-cyclohexyl-dodecanoic acid urea, CUDA). The samples were mixed with a Vortex mixer for 2 min, centrifuged at 14000 ×g for 5 min and then transferred to auto sampler vials with 150 μl inserts for LC/MS analysis.
LC-MS/MS analysis of oxylipins was performed using a modified method based on a previous publication (19). An Agilent 1200 SL liquid chromatography series instrument (Agilent Corporation, Palo Alto, CA, USA) with an Agilent Eclipse Plus C18 2.1×150 mm, 1.8 μm column was used for the separation of the oxylipins. The mobile phase A was water with 0.1% acetic acid while the mobile phase B was composed of acetonitrile/methanol (80/15, v/v) and 0.1% acetic acid. Gradient elution was performed at a flow rate of 250 μl/min according to our previous published program (20). The injection volume was 10 μl and the samples were kept at 4°C in an auto sampler. Analytes were detected by negative MRM mode using a 4000 QTrap tandem mass spectrometer (Applied Biosystems Instrument Corporation, Foster City, CA, USA) equipped with an electrospray ionization source (Turbo V). The QTrap was set as follows: curtain gas=20 psi, temperature=500°C, ion source gas 1=50 psi, ion source gas 2=30 psi, collisionally activated dissociation=high, ion spray voltage=−4500 V, declustering potential=−60 V, entrance potential=−10 V. Calibration curves were generated by 10 μl injections of seven standards containing each analyte, internal standard I, and internal standard II for quantification purpose. The limit of detection and limit of quantitation for t-CUPM were 0.5 nmol/l and 1 nmol/l respectively.
Protein extraction and western blot assay. Freshly harvested pancreases were homogenized and lysed in ice-cold RIPA lysis buffer (Santa Cruz). The lysates were separated by centrifugation at 12,000 × g for 5 min at 4°C; the supernatants were collected and aliquoted. All protein concentrations were determined using the Bradford reagent (Thermo Scientific, Chicago, IL, USA). An aliquot (30 μg protein/lane) of the tissue lysate was separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and then the proteins were transferred onto a polyvinylidene fluoride membrane. The primary antibodies included antibodies against c-RAF, MEK, ERK and their phosphorylated forms, and all of these antibodies were from Cell Signaling Technology. The membranes were further incubated with horseradish peroxidase-linked anti-rabbit IgG and anti-biotin antibodies (Cell Signaling Technology). The protein–antibody complexes were detected by using the chemiluminescent substrate according to the manufacturer's instructions and the emitted light captured on X-ray film. Specific protein band intensity was quantified using Image-Pro Plus image analysis software (Media Cybernetics, Inc. Rockville, MD USA).
Statistical analysis. Each analyzed parameter was expressed as the mean±SD, unless otherwise stated. Continuous variables were compared with the Student's t-test, whereas categorical variables were compared with Chi-square test. All statistical tests were two-sided, statistical significance was taken as p<0.05.
Results
Effect of oral administration of t-CUPM on inhibiting pancreatitis in LSL-KrasG12D/Pdx1-Cre mice. According to pharmacokinetics data that t-CUPM (1 mg/kg body weight) resulted in a plasma Cmax greater than 2-fold half-maximal inhibitory concentration (IC50) for c-RAF inhibition, this dose of t-CUPM in drinking fluid was administered to mice for 4 weeks. There was no significant difference of body weight in LSL-KrasG12D/Pdx1-Cre mice with and without t-CUPM treatment. None of the animals with t-CUPM treatment exhibited any observable toxicity or any gross changes in liver, spleen, kidneys, heart, lung or gastrointestinal tract. There were also no differences of the water and food consumption between mice with and those without t-CUPM treatment.
Pancreatic weight and its ratio to body weight is a simple marker of active pancreatitis. LSL-KrasG12D/Pdx1-Cre mice given a single intraperitoneal injection of caerulein displayed a significant increase in pancreatic weight compared to caerulein-treated C57BL/6J wild-type mice (0.23±0.04 g versus 0.14±0.03 g, p<0.05) and pancreas to body weight ratio (0.011±0.003 versus 0.007±0.002, p<0.05). LSL-KrasG12D/Pdx1-Cre mice treated with t-CUPM exhibited a significant decrease of pancreatic weight (0.18±0.03 g, p<0.05) and its ratio to body weight (0.009±0.001, p<0.05). No significant difference in the weight of other organs, including spleen and liver, was observed between wild-type mice and LSL-KrasG12D/Pdx1-Cre mice as well as LSL-KrasG12D/Pdx1-Cre mice treated with t-CUPM.

Morphological analysis of chronic pancreatitis. A: morphological normal pancreatic parenchyma in wild-type mice. B: Extensive chronic pancreatitis in KrasG12D/Pdx-1-Cre (PK) mice. C: Minimal to mild chronic pancreatitis in PK mice treated with trans-4-{4-[3-(4-chloro-3-trifluoromethyl-phenyl)-ureido]-cyclohexyloxy}-pyridine-2-carboxylic acid methylamide (t-CUPM). D: Histogram of chronic pancreatitis index (CPI: the extent of acinar loss, stromal fibrosis and inflammatory cell infiltration), showing a significant decrease of chronic pancreatitis index (CPI) in PK/t-CUPM-treated mice compared to the PK mice. E-G: Trichrome stain highlighted fibroconnective tissue (blue) only in the interlobular areas of a normal pancreas in wild-type mice (E), extensive fibrosis in pancreatic parenchyma of PK mice (F), and much less stromal fibrosis in the pancreas in PK mice with t-CUPM treatment (G). H: Histogram showing a significant decrease of the extent of stromal fibrosis in the pancreas in PK mice treated with t-CUPM. I–K: Myeloperoxidase (MPO)-labeled neutrophils in the pancreas in wild-type mice (I), in PK mice (K) and in PK mice with t-CUPM treatment. L: Histogram showing a significant decrease of MPO-labeled neutrophils infiltration in the pancreas of PK mice treated with t-CUPM. M–O: Lysosome-associated membrane protein 3 (MAC3)-labeled macrophages in the pancreas in wild-type mice (M), in PK mice (N) and in PK mice treated with t-CUPM (O). P: Histogram showing a significant decrease of MAC3-labeled macrophage infiltration in the pancreas in PK mice treated with t-CUPM. Q–S: Amylase immunostaining showing pancreatic acini in wild-type mice (Q), in PK mice (R), and in PK mice treated with t-CUPM (S), T: Histogram showing a significant preserved pancreas acini in PK mice treated with t-CUPM). U-W: Cytokeratin-19 (CK19)-positive ductal cells in the pancreas in wild type mice (U), in PK mice (V), and in PK mice treated with t-CUPM (W). X: Histogram showing a significant difference in PK mice treated with t-CUPM. *Statistically significant difference, p<0.05.

Histopathological analyses of mouse pancreatic intraepithelial neoplasia (mPanIN) lesions and immunohistochemical analyses of proliferation by proliferating cell nuclear antigen (PCNA) labeling, and phosophorylated extracellular signal-regulated kinases (ERK). A-C: Representative images of mPanIN lesion in wild-type mice (A), KrasG12D/Pdx-1-Cre (PK) mice (B) and KrasG12D/Pdx-1-Cre mice treated with trans-4-{4-[3-(4-chloro-3-trifluoromethyl-phenyl)-ureido]-cyclohexyloxy}-pyridine-2-carboxylic acid methylamide (t-CUPM) (PK/t-CUPM) (C). D: Semi-quantitative analysis (histogram) of mPanIN lesions showing a statistically significant decrease of high-grade PanINs in the pancreas of PK mice treated with t-CUPM). E-G: Proliferation as shown by PCNA labeling of pancreatic tissue: in wild-type mice (E), PK mice (F) and PK/t-CUPM mice (G). H: Histogram of semi-quantitative analysis of PCNA-labeled cell proliferation: t-CUPM treatment significantly reduced the percentage of PCNA-labeled cells in PK mice. I-K: phosphorylated extracellular signal-regulated kinase (p-ERK) expression with immunohistochemical staining: no p-ERK expression in wild-type mice (I), high intensity of p-ERK expression in PK mice (J), significantly reduced p-ERK expression in PK mice treated with t-CUPM (K). L: Histogram of semi-quantitative analysis of the intensity of p-ERK expression: Significant reduction of staining intensity of p-ERK in PK/t-CUPM mice. M: Western blot assay of mutant Kras-activated downstream signals in pancreatic tissues: Expression levels of phosphorylated and non-phosphorylated RAF1 proto-oncogene serine/threonine kinase (c-RAF), mitogen-activated protein kinase's kinase (MEK) and extracellular signal-regulated kinases (ERK) in pancreatic tissues, t-CUPM treatment resulted in a significant reduction of the phosphorylation of c-RAF, MEK1/2 and ERK1/2 proteins. *Statistically significant difference, p<0.05. PDAC: Pancreatic ductal adenocarcinoma.
Histological analysis revealed that the pancreases were unremarkable in wild-type mice either administered vehicle drinking fluid or that containing t-CUPM (Figure 1A). LSL-KrasG12D/Pdx1-Cre mice exhibited extensive chronic pancreatitis as multifocal pattern of acinar loss, terminal ductul proliferation, acinar-ductal mucinous metaplasia, stromal fibrosis, and inflammatory cell infiltration (mainly neutrophils, macrophages and lymphocytes), as shown in Figure 1B. A marked decrease of chronic pancreatitis was observed in LSL-KrasG12D/Pdx1-Cre mice treated with t-CUPM (Figure 1C). The chronic pancreatitis was semi-quantitatively analyzed based on the extent of acinar loss, inflammatory cell infiltration and stromal fibrosis. As shown in the graph of Figure 1D, LSL-KrasG12D/Pdx1-Cre mice treated with t-CUPM displayed a significant decrease of overall score of chronic pancreatitis compared to LSL-KrasG12D/Pdx1-Cre control mice (CPI, chronic pancreatitis index which was a sum of scores of the extent of acinar loss, inflammatory cell infiltration and stromal fibrosis (p<0.01).
The Masson trichrome stain highlighted stromal fibrosis. The pancreas in wild-type mice only showed positive trichrome staining (blue) in the interlobular fibroconnective tissue (Figure 1E). Extensive fibrosis in the pancreatic parenchyma was observed in chronic pancreatitis in LSL-KrasG12D/Pdx1-Cre mice (Figure 1F). Markedly decreased stromal fibrosis was observed in LSL-KrasG12D/Pdx1-Cre mice treated with t-CUPM (Figure 1G). Further semi-quantitative analysis based on staining intensity and extent revealed that LSL-KrasG12D/Pdx1-Cre mice treated with t-CUPM had a significantly decreased fibrosis score compared to LSL-KrasG12D/Pdx1-Cre control mice (p<0.05, Figure 1H).
MPO and MAC3 immunohistochemical stains labeled neutrophils and macrophages, respectively. The pancreas in wild-type mice exhibited no or only rare MPO- or MAC3-positive cells (Figure 1I and M). Numerous MPO-positive neutrophils and MAC3-positive macrophages were observed in the pancreas of LSL-KrasG12D/Pdx1-Cre mice (Figure 1J and N), while markedly decreased number of MPO- and MAC3-positive cells were observed in LSL-KrasG12D/Pdx1-Cre mice treated with t-CUPM (Figure 1K and O) (p<0.05).
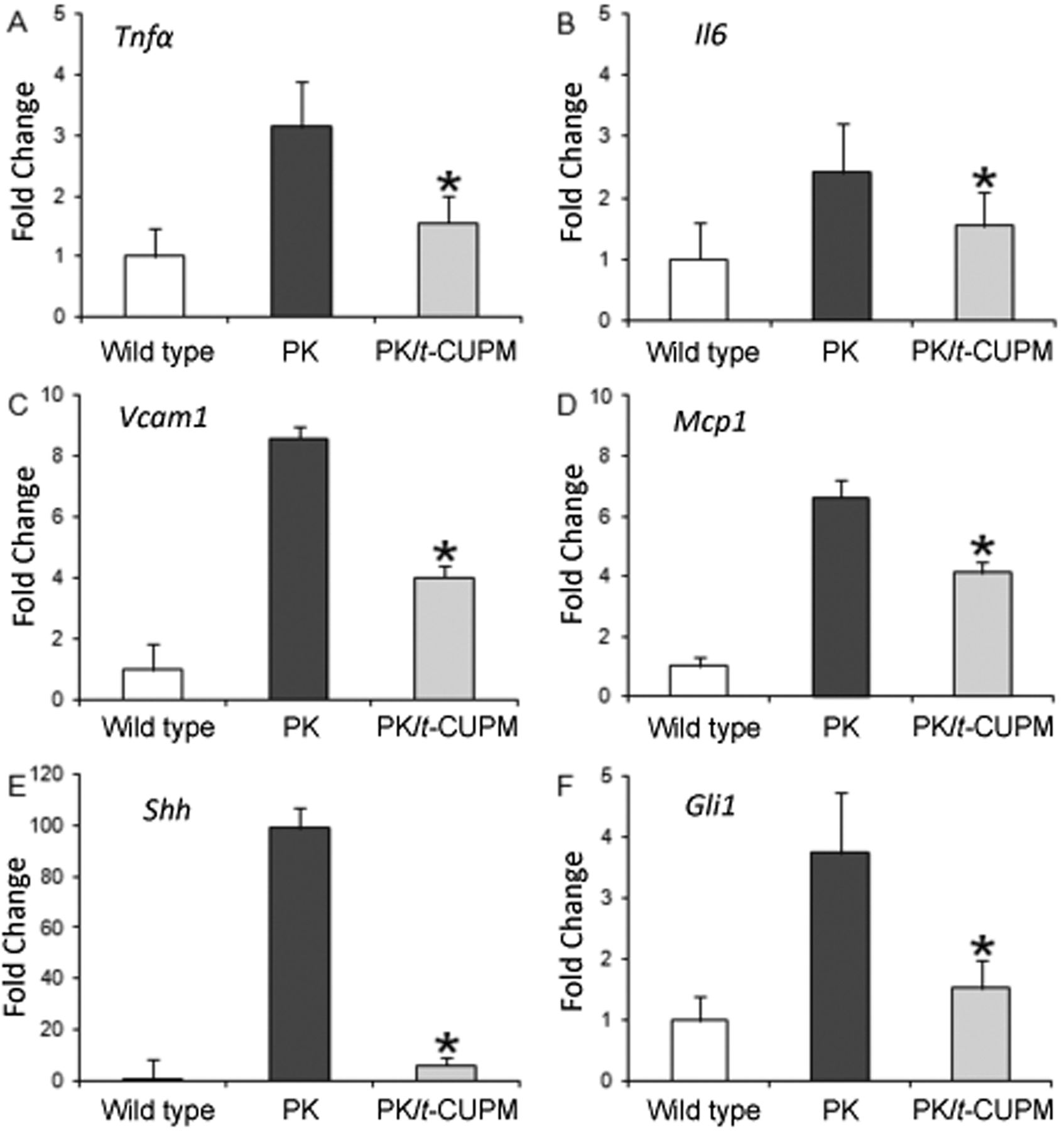
Quantitative real-time polymerase chain reaction analysis of tumor necrosis factor-α (Tnfa) (A), interleukin-6 (Il6) (B), monocyte chemoattractant protein-1 (Mcp1) (C), vascular adhesion molecule 1 (Vcam1) (D), sonic hedgehog (Shh) (E) and Gli transcription factor (Gli) (H) mRNA expression in the pancreas of wild-type mice and KrasG12D/Pdx-1-Cre (PK) mice treated with and without trans-4-{4-[3-(4-chloro-3-trifluoromethyl-phenyl)-ureido]-cyclohexyloxy}-pyridine-2-carboxylic acid methylamide (t-CUPM). *Statistically significant difference, p<0.05.
Acinar loss and ductul proliferation were further analyzed using amylase and cytokeratin 19 (CK19) immunohistochemical approaches. In the pancreas of wild-type mice, amylase-labeled acinar cells were composed of predominant pancreatic parenchyma (>80%), and only approximately 2% were CK19-positive pancreatic ducts (Figure 1Q and U). As shown in Figure 3R and V, chronic pancreatitis in LSL-KrasG12D/Pdx1-Cre mice displayed the marked loss of amylase-positive acinar cells and the increase of CK19-positive ductal cells. Semi-quantitative analysis of the amylase- and CK19-positive area revealed that t-CUPM treatment significantly increased the amylase-positive area of the acini in the pancreas (Figure 1S and 1T), and significantly reduced the CK19-positive area of ductal proliferation (p<0.05).
Inhibition of mPanIN formation and its progression in LSL-KrasG12D/Pdx1-Cre mice treated with t-CUPM. At the age of 12 weeks (8 weeks after caerulein-induced chronic pancreatitis), no pancreatic acinar-ductal mucinous metaplasia or mPanIN lesions were observed in wild-type mice (Figure 2A). All LSL-KrasG12D/Pdx1-Cre mice developed pancreatic acinar-ductal mucinous metaplasia or low-grade mPanIN I lesions, while 8/10 and 3/10 mice developed high-grade mPanIN II and mPanIN III (Figure 2B, left), respectively. One out of 10 mice had an invasive pancreatic adenocarcinoma (Figure 2B, right). t-CUPM treatment resulted in most pancreatic pre-cancerous lesions being acinar-ductal mucinous metaplasia or low-grade mPanIN I lesions (Figure 2C), and a significant decrease of high-grade mPanIN lesions (p<0.05, Figure 2D).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Treatment with trans-4-{4-[3-(4-chloro-3-trifluoromethyl-phenyl)-ureido]-cyclohexyloxy}-pyridine-2-carboxylic acid methylamide (t-CUPM) significantly increased the ratios of epoxyeicosatrienoic acids (EETs) to dihydroxyeicosatrienoic acids (DHETs) and modulated other arachidonic acid metabolic pathways in KrasG12D/Pdx-1-Cre mice (PK) compared to wild-type (WT) mice: A: The ratios of 14(15) EET,11(12)EET, 8(9)EET to their corresponding diol DHETs, and the ratio of total EETs to DHETs. B: Plasma level of prostaglandin E2 (PGE2) and prostaglandin D2 (PGD2) (cyclo-oxygenase-mediated metabolites) and hydroxyicosatetraenoic acid (HETEs, lipoxygenase-mediated metabolites). Each value represents the mean±SD of five mice. *Statistically significant difference, p<0.05.
Inhibition of cell proliferation, and c-RAF, MEK1/2 and ERK1/2 phosphorylation in mice treated with t-CUPM. As shown in Figure 2E-F, PCNA-labeled proliferative cells were rarely observed in the pancreas in wild-type mice, while PCNA-labeled cells were markedly increased in chronic pancreatitis and mPanIN lesions in LSL-KrasG12D/Pdx1-Cre mice. t-CUPM treatment significant reduced the frequency of PCNA-labeled proliferative cells (Figure 2G), and the percentage of PCNA-labeled proliferative cells in total cells counted was significant lower in LSL-KrasG12D/Pdx1-Cre mice treated with t-CUPM (p<0.05, Figure 2H).
Phosphorylation of ERK1/2 protein (p-ERK) as a mutant Kras-activated signal is well-detected immunohistochemically. No such staining was detected in the pancreas in wild-type mice (Figure 2I). mPanIN lesions were strongly stained by the antibody to p-ERK in LSL-KrasG12D/Pdx1-Cre mice (Figure 2J). A marked decrease of staining intensity was seen in LSL-KrasG12D/Pdx1-Cre mice treated with t-CUPM (Figure 2K). The staining intensity of p-ERK was further analyzed semi-quantitatively by Image J software and revealed that the staining intensity was significantly lower in t-CUPM-treated mice (Figure 2L, p<0.05).
Phosphorylation of c-RAF, MEK1/2 and ERK1/2 was further analyzed using western blot approach. As seen in Figure 2M, compared to LSL-KrasG12D/Pdx1-Cre mice (n=3), t-CUPM treatment (n=3) led to a significant reduction of the phosphorylation of c-RAF, MEK1/2 and ERK1/2 proteins, but not of total non-phosphorylated c-RAF, MEK1/2 and ERK1/2 proteins.
Down-regulation of inflammatory cytokines and chemokines as well as Hedgehog pathway in LSL-KrasG12D/Pdx1-Cre mice treated with t-CUPM. Inflammatory cytokines and chemokines were further analyzed using a quantitative real-time PCR approach. The mRNA levels of Tnfa and Il6, and Mcp1, as well as Vcam1 in the pancreatic tissues were significantly increased in the LSL-KrasG12D/Pdx1-Cre mice compared to the wild-type mice. t-CUPM treatment significantly reduced mRNA impression of these inflammatory cytokines and chemokines compared to LSL-KrasG12D/Pdx1-Cre mice (p<0.05, Figure 3)
The Hedgehog pathway plays a crucial role in the fibrosis of chronic pancreatitis. SHH and GLI1 are two of the key molecules in the Hedgehog pathway (21). The mRNA levels of both Shh and Gli1 were significantly increased in the pancreas in LSL-KrasG12D/Pdx-1-Cre mice compared to the wild-type mice. With t-CUPM treatment, the levels of Shh and Gli1 decreased significantly (p<0.05, Figure 3). Analysis of plasma EET profile in mice treated with t-CUPM. The ratio of EETs to DHETs in the plasma are the most commonly used biomarkers for determining sEH inhibition (22). The plasma levels of eicosanoid metabolites were analyzed using an LC-MS/MS method. The ratios of all three sEH-metabolized 8,9-, 11,12- and 14,15-EETs to their corresponding DHETs and total EETs to DHETs were decreased in the LSL-KrasG12D/Pdx1-Cre mice compared to wild-type mice, but these differences did not reach statistical significance. These ratios were dramatically increased in the t-CUPM-treated mice compared to the LSL-KrasG12D/Pdx1-Cre mice, and were restored to levels even greater than that in wild-type mice without pancreatitis (Figure 4A).
To further determine if inhibition of sEH modulates other pathways of eicosanoid metabolism, the products of COX and LOX pathways were analyzed. As shown in Figure 4B, significant increases of PGE2 and D2, and HETEs (8-HETE, 9-HETE, 11-HETE 12-HETE and 15-HETE) were observed in the t-CUPM-treated LSL-KrasG12D/Pdx1-Cre mice compared to LSL-KrasG12D/Pdx1-Cre mice, returning to the levels in wild-type mice without pancreatitis.
Discussion
Inhibition of mutant KRAS-activated ERK pathway and crucial inflammatory event/s is considered the most promising approach for cancer chemoprevention, particularly for pancreatic cancer (23, 24). In the present study, we demonstrated that t-CUPM significantly inhibited chronic pancreatitis and reduced progression of PanIN lesions in a unique genetically engineered pancreatitis-carcinogenesis model in LSL-KrasG12D/Pdx1-Cre mice and further demonstrated the inhibitory effect on phosphorylated MEK-ERK signals and modulation of the eicosanoid metabolic profile. Four-week treatment with t-CUPM did not produce any toxic effect in mice grossly or histopathologically. Our results indicated that t-CUPM as a novel sEH/c-RAF inhibitor has high potential for translation into clinical studies.
PanIN represents proliferative epithelial pre-cancerous lesions in the smaller-caliber pancreatic ducts and ductules which usually originate from chronic pancreatitis (15). Consensus criteria in diagnosis and grading of mPanINs have been established for the models of pancreatic cancer in mice ranging from low grade (PanIN I) to high grade (PanIN II or III) (25). There exist several reports on caerulein-induced pancreatitis and carcinogenesis in the mutant Kras mouse model (26-28), and we have successfully simplified the animal model with only a single intra-peritoneal injection of caerulein to induce and synchronize the development of chronic pancreatitis and mPanINs (14). LSL-KrasG12D/Pdx1-Cre mice given caerulein showed 100% development of acinar-ductal mucinous metaplasia or mPanIN I lesions, 80% mPanIN II, and even notably one out of 10 mice with invasive pancreatic ductal adenocarcinoma at 8 weeks after caerulein treatment. These results indicate that caerulein-induced pancreatitis promotes mutant Kras-initiated carcinogenesis and is a unique mouse model of chronic pancreatitis-carcinogenesis.
Mutant Ras impairs intrinsic GTPase activity, leading to persistent activation of the RAF/MEK/ERK pathway, resulting in cell proliferation and immortalization (29). Using a unique mutant Kras-initiated and pancreatitis-enhanced carcinogenesis in LSL-KrasG12D/Pdx1-Cre mice, we have demonstrated that t-CUPM significantly inhibits cell proliferation, progression of mPanIN lesions, and mutant Kras-activated phosphorylation of MEK1/2 and ERK1/2. These results indicate t-CUPM has high potential for translation into a clinical trial on inhibition of Kras-initiated carcinogenesis.
sEH plays a critical role in regulatory cascades influenced by epoxide-containing lipids. The endogenous sEH substrates are anti-inflammatory EETs, including 8,9-, 11,12- and 14,15-EET (8, 11). Epoxide hydrolysis not only eliminates the biological activity of EETs, but also produces pro-inflammatory dehydro metabolites (30). The omic approach with an LC-MS/MS method to analyze endogenous lipid chemical mediators derived from arachidonic acid provides much more insight than can be obtained from monitoring a single metabolite such as EET. The ratio of lipid epoxides to diols is a simple biomarker for assessing the effects of sEH inhibitor (19, 22, 31). The enzyme activity assay showed that t-CUPM is a most potent sEH inhibitor with an IC50 of 0.5±0.2 nM (12). LC-MS/MS analysis revealed that all three key anti-inflammatory EETs (8,9-, 11,12- and 14,15-EET) were significantly stabilized by t-CUPM in mice with pancreatitis and significantly increased the EET to DHET ratio in the plasma, which further confirmed sEH inhibition in vivo. In addition, our data further revealed that the pattern of modulation of eicosanoid metabolism (including sEH, COX2 and LOX pathways) by t-CUPM treatment appeared to shift from an inflammatory pattern (generally initiating and propagating inflammation in LSL-KrasG12D/Pdx1-Cre mice with pancreatitis) to a pattern of resolution of inflammation (pattern in wild-type mice); in particularly, increased levels of 8-, 9-, 11-, and 15-HETEs may further enhance the anti-inflammatory and anti-carcinogenesis effects (32, 33). Furthermore, inhibition of c-RAF and cell proliferation by t-CUPM indicates that PGE2 elevation caused by sEH inhibition may not be a concern.
Chronic pancreatitis is a complex chronic active inflammatory process in which infiltrating active inflammatory cells (including neutrophils and macrophages) play a crucial role in causing tissue injury (acinar loss) and fibrosis, either through active inflammatory cell-induced tissue damage or via producing oxidative free radicals, inflammatory mediators or cytokines. Our results showed that t-CUPM treatment resulted in a significant decrease of the numbers of neutrophils and macrophages in the pancreas, that was paralleled with a significant decrease of pancreatic injury, as measured by amylase and CK-19 immunohistochemistry. It has been reported that anti-inflammatory action of EETs or sEH inhibitor is mainly through reducing TNF-α-induced VCAM expression and inflammatory cell infiltration, as well as inhibiting NF-κB and Iκκ kinase activities (8). TNFα and IL6 are the best-characterized pro-inflammatory and pro-tumorigenic cytokines that are mainly secreted by macrophages (34-37). MCP1 is a chemokine that recruits monocytes, memory T-cells, and dendritic cells to sites of tissue injury, infection, and inflammation (38). VCAM1 is expressed on both large and small blood vessels after endothelial cell stimulation by cytokines such as TNFα and IL6, and mediates the adhesion of neutrophils, lymphocytes, monocytes, eosinophils, and basophils to vascular endothelium (39). These inflammatory mediators play an important role in inflammatory cell infiltration in inflammation. In our experiment, the production of these inflammatory cytokines or mediators in the pancreas of LSL-KrasG12D/Pdx1-Cre mice was significantly inhibited by t-CUPM treatment. The activation of Shh signals is closely associated with fibrosis/desmoplasia in chronic pancreatitis and pancreatic cancer (40), and mutant Kras is involved in the activation of the hedgehog/GLI1 pathway (41). The mice treated with t-CUPM showed a significant down-regulation of Shh and Gli1. Our results imply that t-CUPM attenuates pancreatitis, particularly by inhibiting inflammatory cell infiltration and fibrosis probably via stabilizing EETs by sEH inhibition, further reducing these key inflammatory mediators and cytokines and suppressing SHH and GLI1.
In summary, with a reasonable oral bioavailability and dual inhibitory activities of sEH and c-RAF, t-CUPM is a very promising agent for inhibiting chronic pancreatitis and PanIN formation, and will have a high potential to translate into clinical trials for preventing and treating pancreatic cancer.
Acknowledgements
This study was supported by NIH R01CA164041 and R01CA172431 to Dr. Guang-Yu Yang.
- Received October 7, 2015.
- Revision received November 7, 2015.
- Accepted November 23, 2015.
- Copyright© 2016 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved