

Abstract
Gallic acid (GA), a phenolic compound naturally present in plants, used as an antioxidant additive in food and in the pharmaceutical industry, may have cancer chemopreventive properties. In the present study, we investigated whether GA induced DNA damage and affected DNA repair-associated protein expression in human oral cancer SCC-4 cells. Flow cytometry assays were used to measure total viable cells and results indicated that GA decreased viable cells dose-dependently. The comet assay and 4’,6-Diamidino-2-phenylindole dihydrochloride (DAPI) staining were used to measure DNA damage, as well as condensation and it was shown that GA induced DNA damage (comet tail) and DNA condensation in a dose-dependent manner. DNA gel electrophoresis was used to examine DNA fragmentation and we found that GA induced DNA ladder (fragmentation). Using western blotting it was shown that GA inhibited the protein expressions of MDC1, O6-methylguanine-DNA methyltransferase (MGMT), p-H2A.X, p53, DNA-dependent serine/threonine protein kinase (DNA-PK) and 14-3-3 proteins sigma (14-3-3σ) but increased p-p53, phosphate-ataxia-telangiectasia (p-H2A.X) and ataxia telangiectasia mutated and Rad3-related (p-ATR), phosphate-ataxia telangiectasia mutated (p-ATM) and breast cancer susceptibility protein 1 (BRCA1) in a 24-h treatment. The protein translocation was examined by confocal laser microscopy and results indicated that GA increased the levels of p-H2A.X, MDC1 and p-p53 in SCC-4 cells. In conclusion, we found that GA-induced cell death may proceed through the induced DNA damage and suppressed DNA repair-associated protein expression in SCC-4 cells.
- Gallic acid (GA)
- DNA damage
- Comet assay
- DNA repair
- human oral cancer SCC-4 cells
It is well-known that maintenance of genomic integrity is vital for the survival and proper function of a cell. It has been reported that DNA damage contributes to the mechanisms of aging and disease (1). DNA damage, inhibition of the DNA repair system, disturbances of cell-cycle distribution and induction of apoptosis are important functions for anticancer drugs (2-4). In cancer chemotherapy, an agent inducing DNA damage and leading cancerous cells to death is of pivotal importance (2, 5), with the induction of reactive oxygen species (ROS) being one of the major factors for causing DNA damage in target cells (6, 7). Furthermore, it has been reported that oncological treatments may generate additional DNA damage and that DNA damage repair has been recognized to be important in both carcinogenesis and cancer treatment (8). It is well-known that cells devote system resources in order to maintain genome integrity and evolve elaborate systems to repair damaged DNA (9).
Gallic acid (GA, 3,4,5-trihydroxy benzoic acid) is an endogenous plant polyphenol naturally found in plants, such as gallnuts, sumac, tea, grapes, berries, oak bark and others. GA has been shown to exhibit antioxidant, anti-inflammatory and anticancer activities (10). Studies have shown that GA protects human cells against oxidative damage (11-13) and that it also exerts anticancer activities in many human cancer cell lines (14-20). GA has also been shown to exert an anti-metastatic effect on human gastric cancer cells (21), A375.S2 human melanoma cells (22), mouse melanoma B16F10 cells (23) and human osteosarcoma U-2 OS cells (24).
Furthermore, it has been reported that, at low concentrations, GA is a potent antioxidant that can prevent oxidative damage to cellular DNA; however, at higher concentrations GA may induce cellular DNA damage (25). Earlier studies of our group have showed that GA caused DNA damage and inhibited DNA repair gene expression in human prostate cancer PC-3 cells (26). However, there is no report to show that GA induces DNA damage and affects DNA repair-associated protein expression in human oral cancer cells. Therefore, in the present study, we measured the effect of GA on DNA damage and repair-associated protein expression in human oral cancer SCC-3 cells.
Materials and Methods
Chemicals and reagents. Gallic acid (GA), dimethyl sulfoxide (DMSO), propidium iodide (PI), trypsin-EDTA, anti-p-ATM, anti-p53, anti-p-p53, anti-DNA-PK and anti-MGMT were purchased from Sigma Chemical (St. Louis, MO, USA). DMEM medium, fetal bovine serum (FBS), L-glutamine and penicillin-streptomycin were purchased from GIBCO®/Invitrogen Life Technologies (Carlsbad, CA, USA). Anti-p-ATR was purchased from Cell Signaling (Danvers, MA, USA), anti-14-3-3σ was purchased from Merck (address) and anti-DNA-PK was purchased from Calbiochem (San Diego, CA, USA).
SCC-4 cell culture. The SCC-4 human oral cell line was obtained from the Food Industry Research and Development Institute (Hsinchu, Taiwan). Cells were immediately cultured in 75-cm2 tissue culture flasks contain 90% Dulbecco's minimal essential medium (DMEM) with 10% FBS and 2 mM L-glutamine containing antibiotics (100 Units/ml penicillin and 100 μg/ml streptomycin) in a humidified atmosphere of 5% CO2, 95% air at 37°C as previously described (26).
Cellular viability. The SCC-4 cells (1×105 cells/well) were placed in 12-well plates for 24 h and then were incubated with 0, 100, 200, 300, 400 and 500 μM of GA for 48 h. Cells from each treatment were collected, centrifuged, washed and stained with PI (5 μg/ml) in phosphate-buffered saline (PBS). Subsequently, the percentage of viable cells was analyzed by flow cytometry (Becton-Dickinson, San Jose, CA, USA), as described previously (26).

Gallic acid (GA) decreased the viability of human oral cancer SCC-4 cells. Cells (1×105cells/well) were placed in 24-well plates and then incubated with GA at 0, 50, 100, 200, 300, 400 and 500 μM for 48 h. All samples were stained with propidium iodide (PI) (5 μg/ml) and analyzed by flow cytometry, as previously described. *p<0.05 was considered significant.
Comet assay (single cell gel electrophoresis). DNA damage was examined by the comet assay. SCC-4 cells (1×105 cells/well) were placed in 12-well plates and then treated with 0, 100, 200 and 300 μM of GA for 48 h in DMEM medium. Briefly, after treatment with GA, aliquots of 105 cells were collected for evaluating cellular DNA damage by using the Comet assay as described previously (25). After electrophoresis, DNA was visualized by ethidium bromide (EtBr) staining and examined and photographed using a fluorescence microscope at 200×. Then the percentage of tail DNA was evaluated as described previously (26). Each result represents measurements from three independent experiments.
4’,6-diamidino-2-phenylindole dihydrochloride (DAPI) staining. SCC-4 cells (1×105 cells/well) were placed in 12-well plates and then treated with 0, 100, 200 and 300 μM of GA for 48 h. The cells were fixed with 3.7% formaldehyde in PBS for 10 min at room temperature followed by two PBS washes. Cells were stained using DAPI solution as previously described (26). Then, cells were examined and photographed using a fluorescence microscope at 200×.
DNA gel electrophoresis. SCC-4 cells (1×106 cells/well) were treated with GA (0, 100, 200 and 300 μM) for 48 h. Then, cells were collected and lysed in 400 μl of ice-cold lysis buffer (containing 50 mM Tris–HCl, pH 7.5, 10 mM EDTA and 0.3% Triton X-100) for 30 min. Cells were centrifuged and RNase A (100 μg/ml) was added to the supernatant and incubated at 50°C for 30 min. Subsequently, proteinase K (200 μg/ml) was added and the mixture was incubated at 50°C for 1 h. Phenol/chloroform was used to extract DNA that was precipitated at −20°C with ethanol/sodium acetate, as described previously (27). Isolated DNA was quantitated and electrophoresed on a 1.5% agarose gel containing 0.1 μg/ml EtBr. Gels were examined as previously described (27).

GA-induced DNA damage in SCC-4 cells was examined by the comet assay. Cells (1×105cells/well) were placed in 12-well plates and then incubated with GA (0, 100, 200 and 300 μM) for 48 h. DNA damage was determined by the comet assay as described in Materials and Methods. A: representative picture of comet assay; B: Comet length (folds of control). Arrow showing the comet tail (DNA damage). ***p<0.001 were considered significant.
Western blotting. SCC-4 cells (1×106 cells/well) were maintained in a 10 cm dish and treated with 0, 100, 200 and 300 μM of GA for 48 h. Cells were harvested, lysed and total proteins from each treatment were determined with the Bio-Rad assay Kit (Bio-Rad, Hercules, CA, USA) as described previously (26, 27). An equal amount of protein from each treatment was electrophoresed on 10% sodium dodecyl sulfate-polyacrylamide gel (SDS-PAGE) and thereafter transferred from SDS-PAGE to a nitrocellulose membrane (Amersham Pharmacia Biotech, Buckinghamshire, UK). The membranes were stained by primary antibodies, such as anti-MDC1, MGMT, p-H2A.X, p-p53, p53, DNA-PK, p-ATR, p-ATM, 13-3-3σ and BRCA1 overnight at 4°C. Then, the membranes were washed and stained with horseradish peroxidase-conjugated anti-mouse secondary antibody (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA) for 1 h at room temperature. Finally, the membranes were visualized with a chemiluminescent detection system and the protein level was determined as described by the manufacturer (26, 27).

GA-induced DNA condension in SCC-4 cells was examined by 4,6-diamidino-2-phenylindole dihydrochloride (DAPI) staining. Cells (1×105 cells/well) were placed in 12-well plates for 24 h and treated with 0, 100, 200 and 300 μM of GA for 48 h. Then, cells were DAPI stained as described in Materials and Methods. Cells were examined and photographed using a fluorescence microscope at 200×. ***p<0.001 were considered significant.
Confocal laser microscopy. SCC-4 cells (5×104 cells/well) were placed on 4-well chamber slides and incubated with GA at the final concentration (0 and 300 μM) for 48 h. After incubation, cells were rinsed with PBS and fixed in 4% formaldehyde in PBS for 15 min at room temperature, followed by 0.3% Triton-X 100 in PBS for 1 h to permeabilize the cells at room temperature. After washing with PBS twice and blocking (5% BSA in PBS, 20 min) were completed, the cells were individually stained overnight with anti-p-H2A.X, anti-p-p53 and anti-MDC1 that contained green fluorescence. After washed twice with PBS and stained with FITC-conjugated goat anti-mouse IgG (secondary antibody), mitotracker (red fluorescence) staining the nuclein followed. The samples were mounted, viewed and photomicrographed under a Leica TCS SP2 Confocal Spectral Microscope as described previously (26, 27).
Statistical analysis. All data are presented as means±standard deviation (S.D.). The comparisons between GA-treated and untreated (control) groups were performed by the Student's t-test. For all comparisons, differences were considered statistically significant at p<0.05.
Results
GA decreases the percentage of viable SCC-4 cells. In order to examine the effects of GA on SCC-4 cells, we treated cells with 0, 100, 200, 300, 400 and 500 μM of GA for 48 h and the percentage of viable cells was measured by flow cytometry. Results are shown in Figure 1. Statistically significant differences regarding reduced cell viability were found at 100 and 500 μM of GA treatment (p<0.05). The observed GA decrease was dose-dependent.
GA induces DNA damage in SCC-4 cells. To further confirm whether GA decreased the percentage of viable cells via the induction of DNA damage, we performed a Comet assay and results are depicted in Figure 2A and B. GA induced DNA damage (Comet tail production) in SCC-4 cells in a dose-dependent manner. GA at 100, 200 and 300 μM induced longer comet tail (DNA migration smear) in SCC-4 cells (Figure 2A) when compared to untreated cells. Thus, increasing GA doses leads to a greater DNA damage (longer comet tail) (Figure 2B).

GA induced DNA fragmentation in SCC-4 cells. Cells (5×105 cells/well) were placed in 12-well plates for 24 h and treated with 0, 100, 200 and 300 μM of GA for 48 h. DNA was extracted and DNA gel electrophoresis was performed. The characteristic pattern of apoptotic inter nucleosomal DNA cleavage (DNA fragmentation) was, finally, visualized.

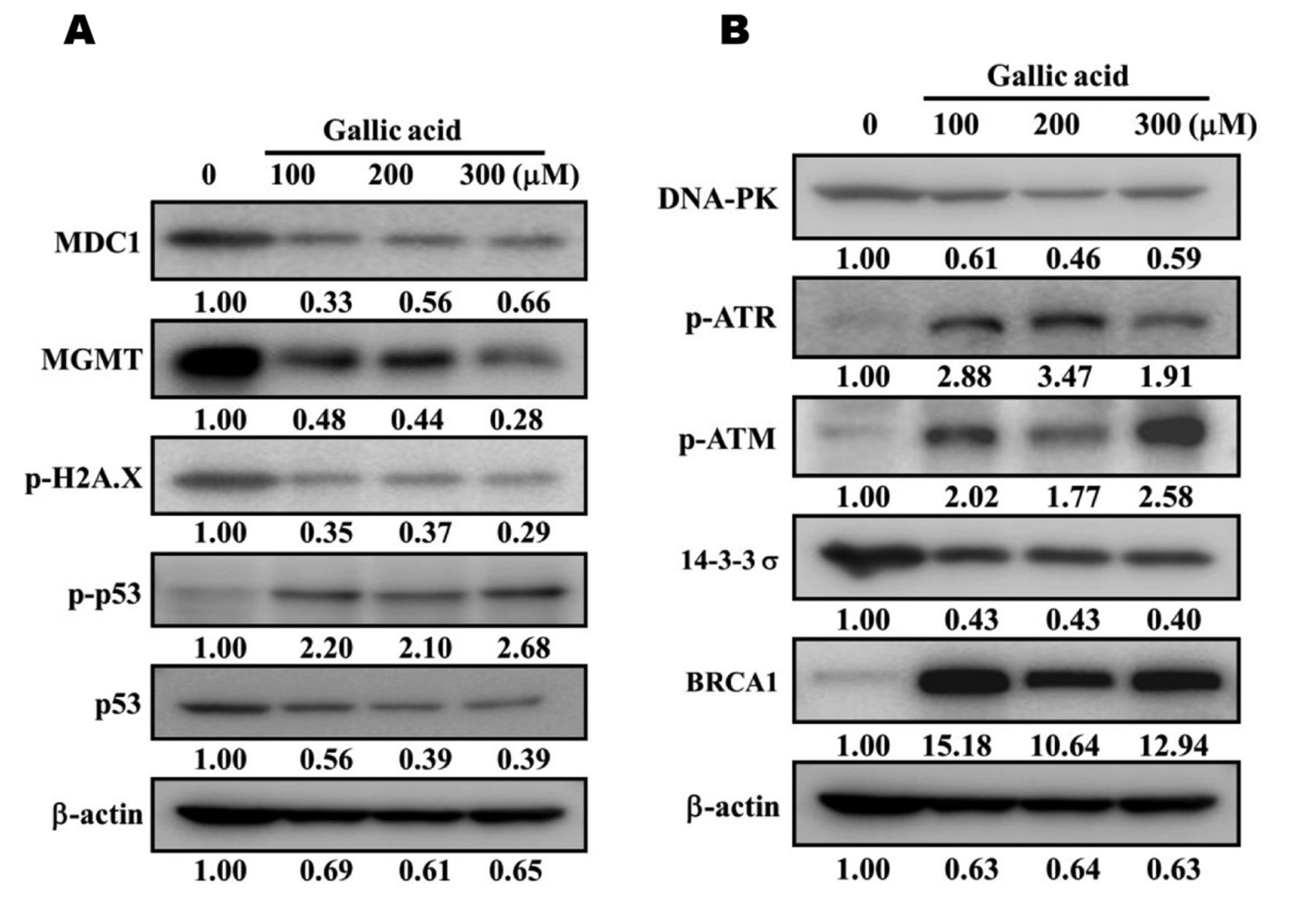
GA affected the protein expression associated with DNA damage and repair in SCC-4 cells. Cells (5×105 cells/well) were placed in 12-well plates and then incubated with 0, 100, 200 and 300 μM of GA for 48 h. Total proteins were collected and determined by the Bio-Rad assay Kit. The amounts of proteins from each treatment were measured by SDS-PAGE and immunoblotting as in Materials and Methods. A: MDC1, MGMT, p-H2A.X, p-p53 and p53; B: DNA-PK, p-ATR, p-ATM, 13-3-3σ and BRCA1.

GA-affected protein expression and translocation in SCC-4 cells were examined by confocal laser microscopy. Cells (5×10 4 cells/well) were kept on 4-well chamber slides and incubated with 0 and 300 μM of GA for 48 h. After fixation in 4% formaldehyde in PBS for 15 min and incubation with 0.3% Triton-X 100 in PBS for 1 h, immunostaining was performed as described in Materials and Methods. A: p-H2A.X; B: p-p53; and C: MDC1 were examined and photomicrographed under a Leica TCS SP2 Confocal Spectral Microscope.
GA induces DNA damage and condensation in SCC-4 cells. In order to further confirm the GA-induced DNA damage in SCC-4 cells, cells were exposed to 0, 100, 200 and 300 μM and subsequently stained by DAPI. The results in Figure 3A and B indicate that GA dose-dependently induced DNA condensation in SCC-4 cells, and it was seen that the higher the GA concentration, the higher the DAPI staining intensity (Figure 3A), and the lower the cell number observed (Figure 3B). Of note, control cells displayed nuclei with homogeneous chromatin distribution and light staining by DAPI.
GA induces DNA fragmentation in SCC-4 cells. DNA fragmentation, the hallmark of DNA damage, was also examined in order to validate the above-described results. Thus, SCC-4 cells were exposed to GA (0, 100, 200 and 300 μM) for 48 h and DNA was isolated and subsequently subjected to gel electrophoresis. DNA fragmentation (DNA ladder) was seen in SCC-4 cells treated with 200 and 300 μM of GA (Figure 4).
GA affects DNA damage and repair-associated protein expression in SCC-4 cells. To investigate whether or not GA induced cell death via DNA damage and repair-associated protein expression, SCC-4 cells were treated with 0, 100, 200 and 300 μM of GA for 48 h and expression of proteins was examined by western blotting. As shown in Figure 5A, GA inhibited the protein expressions of MDC1, MGMT, p-H2A.X and p53 (Figure 5A), DNA-PK and 14-3-3σ (Figure 5B) but increased p-p53 (Figure 5A), p-ATR, p-ATM and BRCA1 (Figure 5B).
GA affects p-H2A.X, p-p53 and MDC1 expression and translocation in SCC-4 cells. To examine whether p-H2A.X, p-p53 and MDC1 are involved in the translocation machineries after GA treatment, we used confocal laser microscopy. The results indicated that GA increased the expression of the p-H2A.X (Figure 6A), p-p53 (Figure 6B) and MDC1 (Figure 6C) in SCC-4 cells when compared to control groups.
Discussion
Numerous studies have shown that GA induces cytotoxic effects on human cancer cell lines through cell-cycle arrest and induction of apoptosis; however, the exact molecular mechanisms for GA-induced cell death in human oral cancer cells associated with DNA damage are still unclear. It is well-known that tumor cells, after exposure to chemotherapeutic drugs, go through DNA repair pathways to survive from the action of chemotherapeutics that cause cellular DNA damage. Therefore, analysis of DNA repair pathways could be used as a prognostic and/or predictive tool (28). Herein, we investigated the effects of GA on DNA damage and repair pathways in human oral cancer SCC-4 cells. Cytotoxic studies have indicated that GA dose-dependently induced cell death (Figure 1). GA induced DNA damage (Figure 2A and B) and condensation (Figure 3A and B) in SCC-4 cells, as measured by the Comet assay (29, 30) and the DAPI staining method, respectively; both effects were dose-dependent. This is in agreement with our earlier report showing that GA induced DNA damage in human prostate cancer PC-3 cells (26). DAPI staining on DNA condensation also confirms our earlier studies (26).
It is well-documented that cells after exposure to agents that cause DNA damage go through DNA repair mechanisms to eliminate DNA lesions or to produce new DNA for maintaining survival (31, 32). The ataxia-telangiectasia mutated (ATM) protein, a kinase that forms a central node in the phosphorylation cascade of the DNA damage response, plays a major role in regulating the cellular response to DNA double-strand breaks (2). Furthermore, ATM can be recruited to sites of DNA lesions and forms large protein complexes that initiate damage signaling (33). Results from Figure 5B indicated that GA increased the p-ATM and p-ATR in SCC-4 cells. It has already been reported that agents induce double-stranded DNA breaks in cells that can activate ATM and ATR to maintain genomic integrity (34, 35). We herein confirmed that GA increased p-p53 (Figure 5A), p-ATR, p-ATM and breast cancer susceptibility protein 1 (BRCA1) (Figure 5B) in SCC-4 cells. ATM also can be phosphorylated by p53, H2A.X, BRCA1 for causing DNA repair (34, 36, 37). It has also been reported that agents induce cell DNA damage, which will induce p-p53 expression (38) and that BRCA-1 is also involved in DNA repair and maintenance of genomic stability (39).
Deficiency and failures in DNA damage response mechanisms could lead to increased cell sensitivity to DNA-damaging factors (40). Our results showed that GA inhibited protein expression of MDC1, MGMT, p-H2A.X and p53 (Figure 5A), DNA-PK and 14-3-3σ (Figure 5B) following 48-h treatment. It has been reported that GA induces cell death via cell-cycle arrest and induction of apoptosis in human cancer cells. Herein, we claim that GA induced DNA damage that is involved in cell death; however, further studies are required to delineate the exact mechanism(s) of action. At the DNA break sites, H2A.X represents a critical factor for the efficient accumulation of DNA repair factors. To this end, it was reported that H2A.X-deficient mice will have higher radiosensitivity compared to normal mice (41) and that the activation of ATM and DNA–PK can proceed via these processes (42).
In conclusion, GA induced cell death of SCC-4 cells in vitro through induction of DNA damage and condensation, while it also affected DNA repair protein expression as it inhibited the protein expressions of MDC1, MGMT, p-H2A.X and p53 (Figure 5A), DNA-PK and 14-3-3σ (Figure 5B), but, at the same time, it increased p-p53 (Figure 5A), p-ATR, p-ATM and BRCA1 (Figure 5B). These effects are summarized in Figure 7.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The possible flow chart for GA-induced DNA damage and inhibition of DNA repair-associated protein expression in human oral cancer SCC-4 cells.
Acknowledgements
The present study was supported by the grant NSC98-2320-B039-027-MY3 from the National Science Council, Taipei, Taiwan. Experiments and data analysis were performed, in part, through the use of the Medical Research Core Facilities Center, Office of Research & Development at China medical University, Taichung, Taiwan, R.O.C.
Footnotes
-
* These Authors contributed equally to this study.
- Received October 3, 2014.
- Revision received December 4, 2014.
- Accepted January 13, 2015.
- Copyright© 2015 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved