

Abstract
Aim: This study aimed to investigate the relationship between prognosis after curative hepatectomy and serum methylation signature (SMS), defined by methylation levels of six specific genes (cyclin D2, Ras association (RalGDS/AF-6) domain family member 1, serine peptidase inhibitor Kunitz type 2, cystic fibrosis transmembrane conductance regulator, brain abundant membrane attached signal protein 1, and steroid-5-alpha-reductase alpha polypeptide 2). Patients and Methods: Serum samples were collected preoperatively from 125 patients with hepatocellular carcinoma associated with hepatitis C virus infection who underwent curative hepatectomy. We measured the methylation levels of the preceding six genes. We defined the methylation of three genes or more in the serum as SMS-positive in this study. We investigated the prognosis of SMS-positive patients. Results: SMS-positive patients exhibited significantly shorter disease-free survival (DFS) and overall survival (OS) than SMS-negative patients (p=0.0002 and p<0.0001, respectively). Multivariate analysis showed that SMS positivity was an independent risk factor for shorter DFS (hazard ratio (HR)=2.182; p<0.001) and OS (HR=4.198; p<0.001). Conclusion: SMS is useful as a prognostic predictor in patients with hepatocellular carcinoma after curative hepatectomy.
- HCC
- cell-free DNA
- methylation
- epigenetic
- quantitative MSP
Hepatocellular carcinoma (HCC) develops mainly from chronic liver diseases such as persistent infection with the hepatitis B virus (HBV) and the hepatitis C virus (HCV). Due to the continued increase in the incidence of HCC in many countries, HCC represents a major international health problem (1, 2). Although resection provides one of the best chances for a cure in patients with HCC, data from the International Cooperative Study Group for Hepatocellular Carcinoma show a poor 5-year survival rate of only 31-41%, regardless of ethnicity (3). This modest prognosis is largely attributable to the high rate of intrahepatic recurrence (IHR) of the disease (4, 5). To improve the poor prognosis, it is necessary to identify a simple prognostic marker for HCC that would be valid prior to surgical therapy.
It is well-established that epigenetic inactivation of gene expression linked to aberrant methylation on C–phosphate–G (CpG) islands serves as a fundamental contributor to carcinogenesis and cancer progression (6-8). Since the CpG island methylator phenotype (CIMP), characterized by the simultaneous methylation of multiple genes, was found to be associated with a molecular sub-class of colon cancer (9), the relationship of CIMP status to a sub-class of HCC has been well-documented (10-13). Many recent studies have shown that CIMP status in tumor tissues is associated with up-regulation of telomerase activity, progression, and recurrence of HCC (10-13). However, all those studies used resected tumor tissues to measure the methylated form of several targeted genes, suggesting that their predictors based on tumor tissues do not work prior to surgery (10-13). Our recent study showed that methylation level of the cyclin D2 (CCND2) gene in tumor tissue were consistent with its methylation level in serum in patients with HCV-related HCC (14). This raises one hypothesis that blood (i.e. serum) could serve as an easy-to-use substitute for the assessment of the methylation signature such as CIMP at the primary tumor, and the methylation signature in serum might serve as a prognostic predictor of HCV-related HCC prior to surgery. The purpose of the present study was to investigate the association between the methylation level of several genes in the serum and the metastatic potential of HCV-related HCC, such as early IHR due to intrahepatic metastasis.
Patients and Methods
Patients. The present study enrolled 125 patients with HCV-associated HCC who underwent curative hepatectomy in our Institute between August 1993 and December 2009. The patients were followed up according to the surveillance program described previously (15). The tumor-node-metastasis (TNM) staging system revised by the Liver Cancer Study Group of Japan (LCSGJ) was used for this study. Written informed consent was obtained from all patients prior to their entry into this study. The study protocol was approved in advance by the Institutional Review Board for the Use of Human Subjects at the Yamaguchi University School of Medicine.
DNA extraction and evaluation by quantitative methylation-specific polymerase chain reaction (QMSP) in cell-free DNA in serum. Serum samples were collected preoperatively from patients who had HCC associated with HCV. We examined the serum methylation level of six genes (CCND2, Ras association (RalGDS/AF-6) domain family member 1 (RASSF1A), serine peptidase inhibitor Kunitz type 2 (SPINT2), cystic fibrosis transmembrane conductance regulator (CFTR), brain abundant membrane attached signal protein 1 (BASP1) and steroid-5-alpha-reductase alpha polypeptide 2 (SRD5A2)) in cell-free DNA (cfDNA). The six genes were selected from our DNA database and based on the literature (Figure 1), and judged as suitable for our methylation marker study on the basis of validation with the use of pyrosequencing after bisulfate treatment (15, 16).
DNA was extracted from 0.5 ml of sera using the DNA Extractor SP Kit for serum and plasma (Wako Pure Chemical Industries Ltd., Osaka, Japan). The extracted DNA was treated with bisulfate and quantified. We used the QMSP assay to measure methylated levels of six genes in the DNA extracted from serum (Table I).
For the determination of the methylation level of six genes in serum, the amount of methylated DNA in 5 μl (1 ng) of bisulfate-treated DNA solution was quantified by using a standard curve constructed from simultaneously measured standards made from a dilution series (2000, 1000, 500 and 250 pg/μl) of artificially methylated DNA (CpGenome™ Universal Methylated DNA; Chemicon International Inc., Temecula, CA, USA). Serum methylation levels were calculated as the relative amount of methylated DNA (measured as pg/1 ml serum). Our system was unable to quantify values below 0.2 pg/1 ml serum (14). Thus, serum samples with values greater than 0.2 pg/1 ml serum were considered positive for methylation of genes in the present study.
Statistical analysis. The Chi-square test or Fisher's exact test was used to evaluate statistical differences between two or three categorical variables. We used the Cox proportional hazards model to assess independent factors for disease-free survival (DFS) and overall survival (OS). Eight variables, namely tumor size, tumor differentiation, venous invasion, stage, the presence of liver cirrhosis (LC), alfa-fetoprotein (AFP), prothrombin-induced vitamin K absence II (PIVKA-II), and serum methylation signature (SMS), were entered into a forward stepwise regression model. Each model was tested for goodness of fit by −2 log likelihood and chi-square in each step. For DFS and OS, survival curves were constructed using the Kaplan–Meier method, and statistical significance was determined by the log-rank test. All statistical analyses were performed using SPSS 19 (SPSS; IBM, Chicago, IL, USA) software. Levels of p<0.05 were considered to be statistically significant.
Results
Patients' clinicopathological features associated with genes. Patients sero-positivity for the presence of methylated CFTR and BASP1 were significantly older than the patients who were sero-negative for these genes (p=0.042 and p=0.039, respectively; Table I). Tumor size was significantly larger in patients sero-positive for methylated CCND2, RASSF1A, SPINT2, and CFTR than in those sero-negative (p=0.037, p=0.013, p<0.001 and p=0.028, respectively; Table I). Tumor number was significantly greater in patients sero-positive for methylated BASP1 than in those sero-negative (p=0.05; Table I). Tumor differentiation grade was significantly lower in patients sero-positive for methylated BASP1 (p=0.013; Table I). Patients sero-positive for methylated CCND2 and SRD5A2 had more advanced HCC than did those who were sero-negative (p=0.019 and p=0.029, respectively; Table I). There were no associations between gender or venous invasion and methylation of any of the six genes in serum (Table I).
Patients' clinicopathological features associated with early IHR. Sixty-four (51.2%) out of the 125 study patients demonstrated IHR within two years after curative hepatectomy (Table II). The frequency of early IHR tended to be higher in males than in females (p=0.080; Table II). Patients with early IHR tended to be younger than those without early IHR (p=0.099; Table II). There were no associations between other factors and early IHR (Table II).
Relationship between SMS and early IHR. The frequency of early IHR was significantly higher in patients sero-positive for methylated SPINT2 and SRD5A2 than in those who were sero-negative (p=0.018 and p=0.036, respectively, Table III).
Among the six genes tested, SPINT2 and SRD5A2 displayed high specificity (82.0% and 93.4%) but low sensitivity (37.5% and 20.3%) for early IHR detection (Table III).
Our data showed that in parallel to an increase in the number of methylation-positive genes, the frequency of early IHR became high (Figure 2). Thus, we examined the relationship between number of genes with methylation (n1-6) and early IHR, and evaluated specificity, sensitivity, accuracy and Youden's index (Figure 2, Table III). We found that positivity for methylation of more than three genes achieved the best performance (specificity=67.2%, sensitivity=56.3%, accuracy=62.4% and Youden's index=0.235) in all combinations tested (Table III). Therefore, we defined methylation of more than three genes in the serum as SMS-positive in the present study.
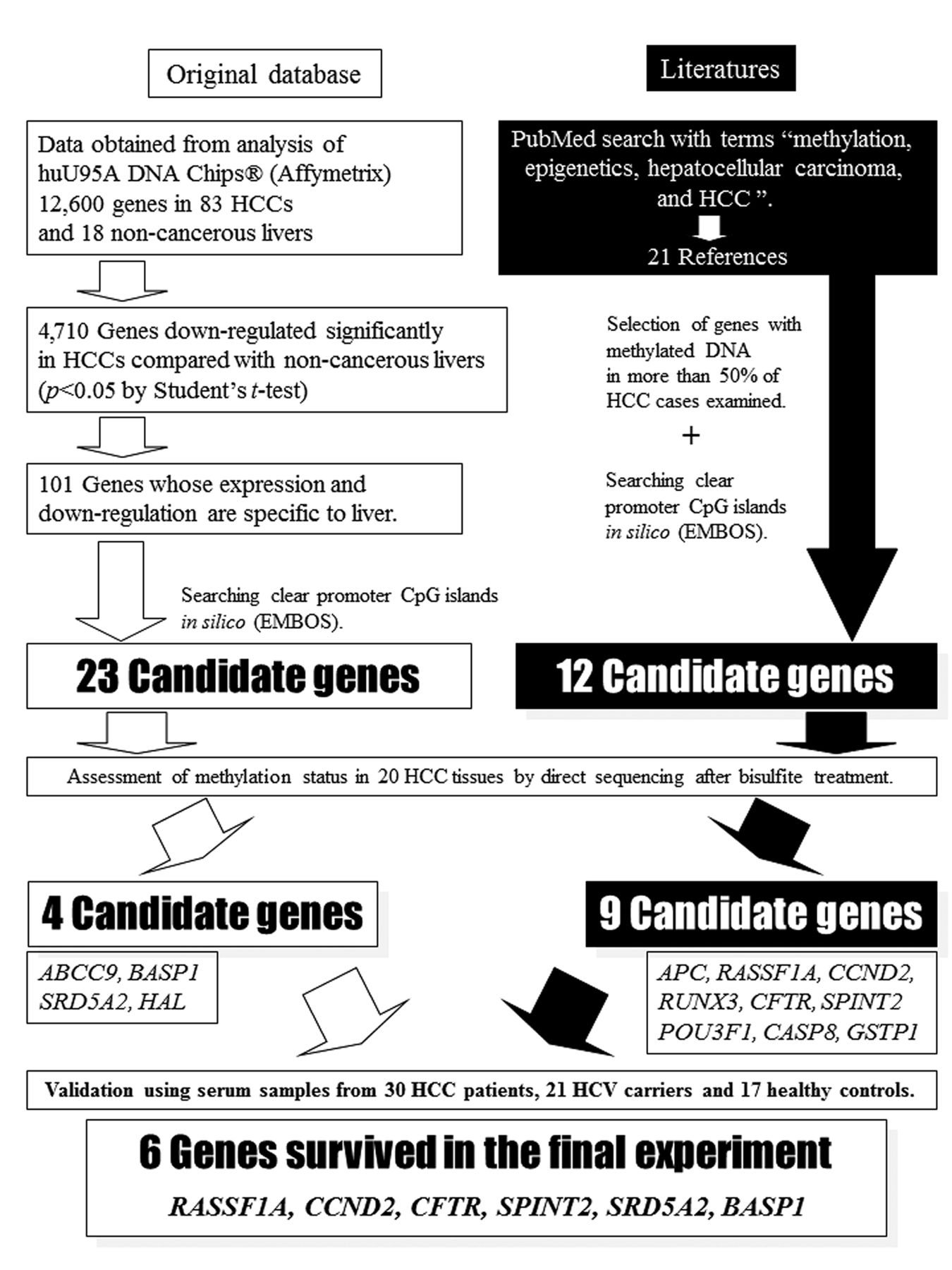
Schema for selection of candidate genes (cyclin D2 (CCND2), Ras association (RalGDS/AF-6) domain family member 1 (RASSF1A), serine peptidase inhibitor Kunitz type 2 (SPINT2), cystic fibrosis transmembrane conductance regulator (CFTR), brain abundant membrane attached signal protein 1 (BASP1) and steroid-5-alpha-reductase alpha polypeptide 2 (SRD5A2)). We examined the serum methylation level of six genes (RASSF1A, CCND2, CFTR, BASP1, SPINT2, and SRD5A2) in cell-free DNA that were selected from our DNA database and the literature.
SMS was positive in 56 (44.8%) out of the 125 study patients. Tumor size was significantly larger in SMS-positive patients than in SMS-negative patients (p=0.005; Table IV). There were no associations between SMS positivity and other clinicopathological features such as patient gender or age, state of the liver, number of tumors, tumor differentiation, venous invasion, stage, AFP, or PIVKA-II (Table IV).
Patient clinicopathological features associated with six genes.
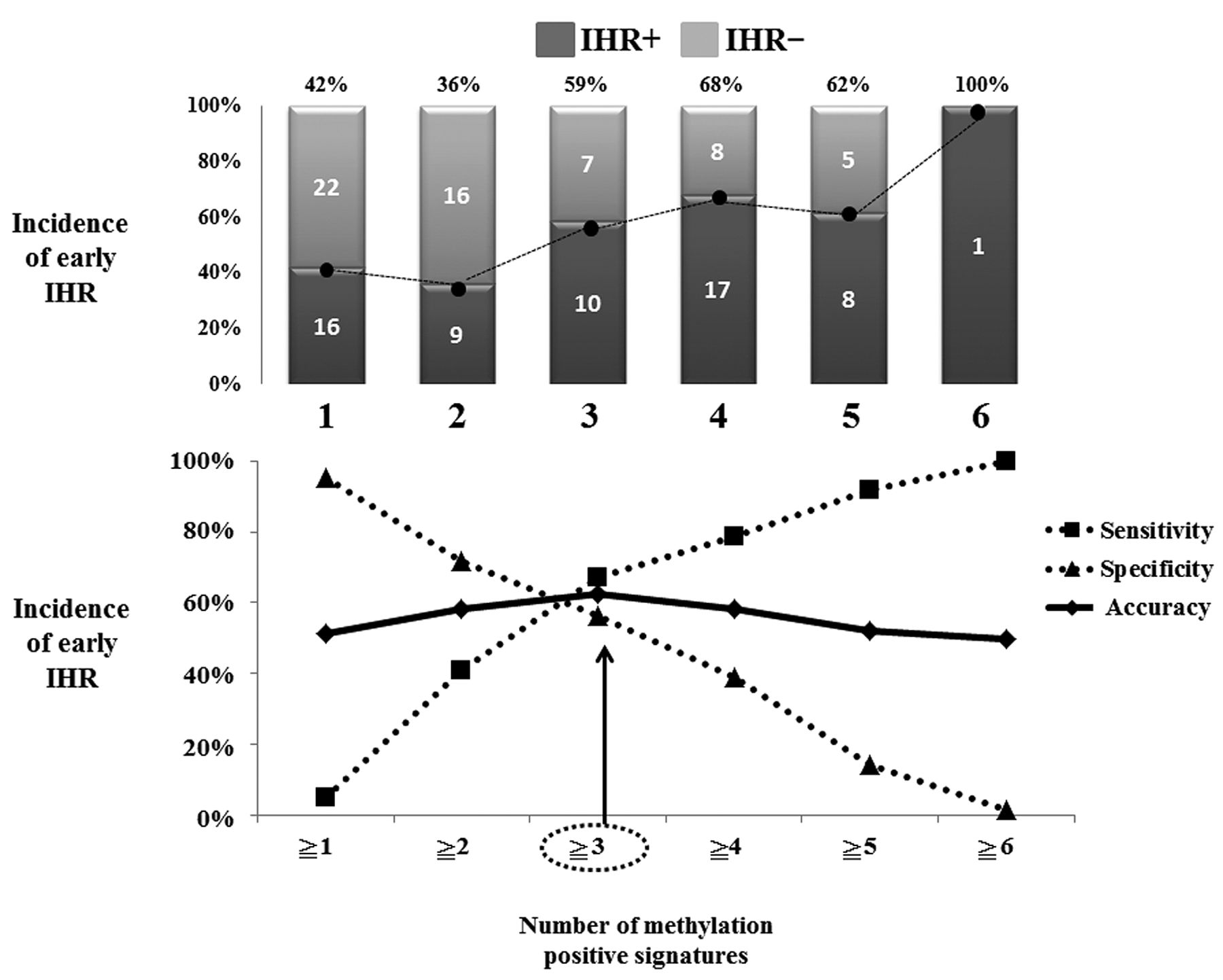
Relationship of serum methylation signature (SMS) and early intrahepatic recurrence (IHR). Our data showed that in parallel to an increase in the number of methylation-positive genes, the incidence of early IHR became high. We examined the relationship between the number of methylated genes (n≥1-6) and early IHR, and evaluated their specificity, sensitivity, and accuracy in predicting IHR. We found that methylation positivity of more than three genes achieved the best performance (specificity=67.2%, sensitivity=56.3%, and accuracy=62.4%) in all combinations tested. Therefore, we defined methylation of more than three genes in the serum as SMS-positive in the present study.
Although the performance of SMS in detecting early IHR was better than the single use of six genes, the individual six genes had, a Youden's index (sensitivity+specificity–1) greater than 0.5 for detection of early IHR in our cohort (Table III).
Relationship between SMS and prognosis. Patients sero-positive for methylated CCND2 and SPINT2 had significantly shorter DFS than those who were sero-negative (p=0.028 and p=0.001, respectively; Figure 3). Patients sero-positive for methylated CCND, RASSF1A, BASP1, and SPINT2 had significantly shorter OS than those who were sero-negative (p=0.002, p=0.007, p=0.030, and p<0.001, respectively; Figure 4). SMS-positive patients had significantly shorter DFS and OS than SMS-negative patients (p=0.0002 and p<0.0001, respectively; Figure 5).
We used the Cox proportional hazards model to assess independent factors for DFS and OS. Fourteen variables, namely tumor size, tumor differentiation, venous invasion, stage, the presence of liver cirrhosis (LC), AFP, PIVKA-II, the six genes used in the present study, and SMS were entered into a forward stepwise regression model. The results showed that SMS was an independent risk factor for both DFS [hazard ratio (HR)=2.182; p<0.001; Table V) and OS (HR=4.198; p<0.001; Table V).
Discussion
Detection and measurement of circulating cfDNA specific for malignancies has opened new opportunities in predictive oncology (17, 18). In a genome-wide search using DNA array data, our recent study used a QMSP technique to identify two unique genes (BASP1 and SRD5A2) for which promoter methylation is specific for small HCC associated with HCV infection (19). Moreover, we found that four genes (RASSF1A, SPINT2, CCND2, and CFTR) were exclusively methylated in early HCC tissues (16). We have already reported that methylated CCND2 in the serum serves as a predictor of prognosis in HCC after curative hepatectomy (14). The aim of the present study was to develop a more accurate prognostic predictor for patient outcome after curative hepatectomy using methylation of these six genes preoperatively.

Disease-free survival (DFS) according to six genes. Patients who were sero-positive for methylated cyclin D2 (CCND2) and serine peptidase inhibitor Kunitz type 2 (SPINT2) exhibited significantly shorter DFS than patients who were sero-negative (p=0.028 and p=0.001, respectively).
In the present study, the frequency of early IHR was significantly higher in patients who were sero-positive for methylated SPINT2 and SRD5A2 than in those who were sero-negative for these genes. Among the six genes tested, SPINT2 and SRD5A2 displayed high sensitivities (82.0% and 93.4%) but low specificities (37.5% and 20.3%) for early IHR detection.
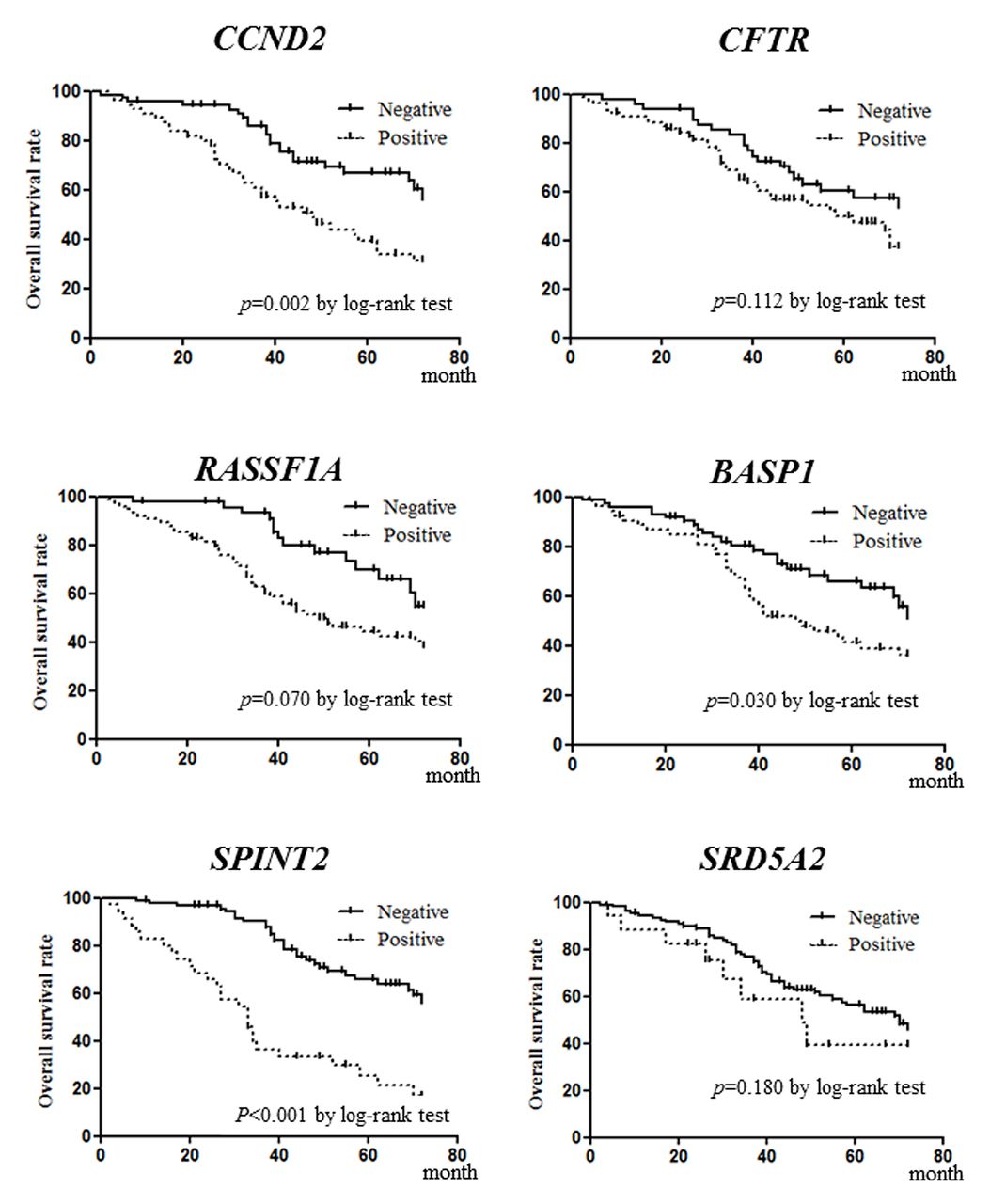
Overall survival (OS) according to six genes. Patients who were sero-positive for methylated cyclin D2 (CCND2), Ras association (RalGDS/AF-6) domain family member 1 (RASSF1A), brain abundant membrane attached signal protein 1 (BASP1) and serine peptidase inhibitor Kunitz type 2 (SPINT2) exhibited significantly shorter OS than patients sero-negative for methylation of these genes (p=0.002, p=0.007, p=0.030 and p<0.001, respectively).
There are limitations in the use of a single marker for providing an accurate prediction of the outcome of patients with cancer because cancer progression needs various genomic and epigenomic changes (20). To resolve these issues, we identified a new biological sub-class linked to outcome defined by methylation signature. We examined the combinations of genetic number to use statistically and evaluated the combinations using Youden's index. Thus, we defined patients with methylation of more than three genes in the serum as SMS-positive in this study. Although using the SMS to detect early IHR was better than the single use of six genes individually, the individual six genes had a Youden's index of more than 0.5 for early IHR detection in our cohort. It was necessary to combine SMS with other clinical parameters such as gender or PIVKA II in order to obtain superior results.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Disease-free survival (DFS) and overall survival (OS) in HCC patients according to the serum methylation signature (SMS) defined by six methylated genes. Among the 125 study patients who underwent curative surgery, 56 were positive for SMS and the remaining 69 patients were negative. SMS-positive patients exhibited significantly shorter DFS and OS than SMS-negative patients (p=0.0002 and p<0.0001, respectively).
However, SMS positivity was found to have clinical importance in patient's prognosis. SMS-positive patients had significantly shorter DFS and OS than SMS-negative patients. Among several clinical factors, SMS-positivity was found to be an independent risk factor for both shorter DFS and shorter OS.
We defined SMS as a CpG island methylator phenotype (CIMP). Since the CIMP has been associated with a molecular subclass of colon cancer (9), the relationship of CIMP status to a sub-class of HCC has been well documented (10-13). Toyota et al. reported that in patients with gastric cancer, CIMP positivity was defined as positive methylation of more than three genes at a primary tumor; and CIMP-positive patients had a poor prognosis (21). Many differences in the definition of CIMP (i.e. different CpG positions or different genes tested, or cut-off value for definition) exist among the studies that have reported the relationship between CIMP status in tumor tissues and prognosis or recurrence of HCC (10-13). Indeed, among the genes used in the present study, five genes other than RASSF1A were not listed as CIMP-related genes for HCC in other studies (10-13). It has also been reported that P16 and glutathione S-transferase pi 1 (GSTP1) are associated with HCC (10-13). Therefore, we preliminarily analyzed P16 and GSTP1, which have been examined in previous CIMP studies with respect to HCC (10-13). However, these genes were not methylated in a cancer-specific manner in the cohort of only HCV-infected patients (10-13). In addition, there were also differences in sample backgrounds (i.e. different races or different hepatitis virus types). A previous molecular profiling study (22) revealed that HBV and HCV cause HCC via different carcinogenetic pathways. Iizuka et al. have reported that there was a significant difference in postoperative clinical course (i.e. outcome) between HBV- and HCV-related HCCs (23). In evaluating a new prognostic marker, it is crucial to minimize the bias caused by distinct agents such as hepatitis viruses. Thus, we investigated whether hypermethylation of six genes in DNA isolated from serum was associated with early IHR or the metastatic potential of HCC in a cohort consisting only of HCV-infected patients.
Patient clinicopathological features associated with early intrahepatic recurrence (IHR).
Relationship between early intrahepatic recurrence (IHR) and methylation signature, and sensitivity, specificity and accuracy.
Bruix and Sherman reported a remarkable advancement in the treatment and diagnosis of HCC (24). However, patients with HCC may have a generally poor prognosis due to their high rate of early IHR caused by intrahepatic spread of cancer cells, even when curative hepatectomy is performed (3, 4, 25, 26). A great deal of effort has been devoted to the development of predictive systems for early IHR. Many genome-wide studies using high-tech array systems (27-30) have raised the possibility of accurately predicting early IHR in patients with HCC. However, a shortcoming of this approach is that such DNA array systems are high in cost, generate unstable information, and require a tissue sample as a source for molecular profiling, raising the issues of preoperative and daily risk assessment in early IHR. In this regard, our serological epigenetic marker may allow for risk evaluation of early IHR or recurrence preoperatively in the setting of everyday clinical practice.
The present study shows the clinical benefit of analysis of the SMS for evaluating the outcome of patients with HCV-related HCC prior to surgical therapy. From the stand-point of everyday clinical use, the development of predictive systems must enable the accurate detection of patient risk for early IHR. Although further studies are needed to correctly predict patient outcome, the preoperative SMS is simple and useful as a prognostic tool for patients with HCC with HCV after curative hepatectomy.
Patient clinicopathological features associated with serum methylation signature (SMS).
Independent risk factors for disease-free survival (DFS) and overall survival (OS).
Acknowledgements
This work was supported by JSPS KAKENHI Grant Number 24592002.
Footnotes
-
Conflicts of Interest
The Authors have no conflicts of interest.
- Received September 22, 2014.
- Revision received October 23, 2014.
- Accepted October 27, 2014.
- Copyright© 2015 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved