

Abstract
Background: Intracranial chondrosarcomas comprise 6% of all skull base neoplasms and account for 0.15% of all intracranial tumors. They are potentially fatal, invading the brain and elevating intracranial pressure by progressive enlargement. Diplopia and headache are the most common clinical symptoms. Previous data indicate a particularly aggressive behavior of intracranial chondrosarcomas. Case Report: A 17-year-old female patient presented to the hospital with focal seizures. A magnetic resonance imaging (MRI) scan revealed a brain tumor located in the right meso-temporal region. Total tumor resection, followed by conformal proton therapy was performed. The tumor displayed a chondroid differentiation, and accordingly, a chondrosarcoma was diagnosed. At follow-up investigation two years after the resection of the tumor, the patient was in a good general state of health and no tumor recurrence had occurred. Discussion and Conclusion: Intracranial chondrosarcoma is a differential diagnosis for intracranial tumors of the skull base. State-of-the-art therapy should comprise of surgical resection and adjuvant radiotherapy. Previously published data about intracranial chondrosarcomas indicate the extreme aggressiveness of this tumor entity.
- Chondrosarcoma
- intracranial tumour
- prognosis
Chondrosarcomas are the third most common primary malignant neoplasms of bone (1). Intracranial chondrosarcomas form 6% of all skull base neoplasms and account for 0.15% of all intracranial tumors (2). Four subtypes of chondrosarcoma are known: conventional (hyaline or myxoid), dedifferentiated, clear cell, and mesenchymal (3). The conventional subtype is composed of either hyaline cartilage, myxoid cartilage or a combination of both. Hypercellular hyaline cartilage with atypical chondrocytes within lacunae characterizes the hyaline type of conventional chondrosarcoma. The myxoid type, by contrast, features atypical chondrocytes which do not reside in lacunae, but in a flocculent myxoid matrix (4). Mesenchymal chondrosarcomas usually display anaplastic features (4). They mainly occur at a younger age (10 to 30 years) (5). Tumor markers, such as vimentin, cytokeratin and S-100 help differentiate chondrosarcoma from chordoma. Chordomas do not usually express vimentin, and chondrosarcomas do not feature cytokeratin, as observed on immunohistochemical staining. Both chordomas and chondrosarcomas usually express S-100 (6).
The prognosis of chondrosarcomas depends on the histological grade. According to the World Health Organization, chondrosarcomas are divided into three categories based on their histological grade: grade I: well-differentiated, grade II: moderately-differentiated, and grade III: poorly-differentiated (4). Grade I chondrosarcoma might be differentiated from enchondroma with difficulty, featuring slightly higher cellular density and more cellular atypia compared to enchondroma (7, 8). Grade II chondrosarcoma is more cellular compared to grade I, with a lobulated growth pattern. The tumor cells feature enlarged, irregular and hyperchromatic nuclei. Grade III chondrosarcoma is hypercellular with enlarged and hyperchromatic cell nuclei that may become fusiform. Mitotic figures and necrosis are commonly seen (7, 8). In the literature, the mesenchymal and myxoid subtypes are classified separately from the conventional type I to III graduation (9).
It has been postulated that intracranial chondrosarcomas arise during enchondral ossification of the bones of the skull base (10). Enchondral ossification takes place in many sites of the skull during bone maturation, for example a large part of the petrous bone, parts of the petro-occipital, spheno-occipital and spheno-petrous synchondrosis are formed by enchondral ossification (4). Chondrocytes within residues of enchondral cartilage which are present in these regions might be the origin of chondrosarcomas. Another theory of the origin of intracranial chondrosarcomas suggests primitive and multifunctional mesenchymal cells, involved in embryogenesis of the skull base, as being the source of origin. Other authors assume that intracranial chondrosarcomas arise from metaplastic mature fibroblasts (11, 12).
Intracranial chondrosarcomas are potentially fatal, invading the brain and elevating intracranial pressure by obstructing the cerebrospinal fluid pathways. Diplopia, caused by compression of intracranial nerves (III, IV and VI), visual field defects and headache are the most common symptoms among patients suffering from intracranial chondrosarcomas (13-16). Since complete surgical resection of intracranial and intracerebral chondrosarcomas is often impossible because of the deep location, a combination of surgery and radiotherapy is today's state-of-the-art approach (13-16). In a review on intracranial chondrosarcomas by Bloch et al., 161 patients who received surgery alone were compared to 325 patients who had surgery in addition to postoperative adjuvant radiation therapy (4). The 5-year mortality rate was higher in the group of patients that were treated with surgery alone (25% vs. 9%; p<0.0001) (4).
According to a review by Iyer and colleagues, stereotactic radiosurgery may provide a benefit for patients suffering from intracranial chondrosarcoma, either as primary or adjuvant therapy (17). In the 22 cases of cranial base chondrosarcomas where stereotactic radiosurgery was performed, tumor control rates (meaning near-total extinction of tumor cells after treatment) were 91%, 72%, 72% and 54% at 1, 3, 5 and 10 years, respectively. The overall survival was 95%, 76%, 70% and 56%, respectively (17). The authors found that tumor control was more likely to be achieved by earlier timing of stereotactic radiosurgery after diagnosis, and by multimodal management including also surgical resection (17).
Case Report
A 17-year-old female patient presented to the hospital with focal seizures. A magnetic resonance imaging (MRI) scan revealed a tumor at the skull base (Figure 1). The tumor was located in the right meso-temporal region, originating from the clivus and expanding to the sella turcica.
After the tumor had been removed totally, the patient developed partial insufficiency of the anterior pituitary. This condition resulted in secondary amenorrhea and hypothyreosis. The patient also experienced somatotropin and adrenocorticotropic hormone deficiency, and the latter caused hypocortisolism. Therefore, the patient was treated with hydrocortisone (15 mg per day) and levothyroxine (37.5 μg per day). An estradiol and gestagen preparation was administered, and subsequently the patient gained normal menstruation. Levetiracetam was administered because seizures persisted postoperatively. Due to the operation, ischemic areas were seen subcortically in the fronto-parieto-temporal region, in the caudate nucleus, and in the basal ganglia on the right side, leading to a transient paresis of the left arm, an impaired field of vision on the right side and bilateral anisocoria.
Conformal proton therapy was performed as adjuvant therapy. At a follow-up investigation two years after the resection of the tumor, the patient was in a good general state of health. No recurrent or residual tumor was visible on the MRI scan. The patient's muscle tonus and grip strength were normal, bilaterally. Muscle reflexes were also normal on both sides. No anisocoria was observed and the pupillary light reflexes were normal. Gross motor skills were within normal range, yet a low-grade dysdiadochokinesis was observed. Electroencephalography was normal, with no signs of deceleration or cerebral hyperexcitability. The patient reported no more seizures upon treatmet with levetiracetam in the previous few months. Apart from moderate headache episodes linked to changes in weather conditions, the patient stated having no disabilities. Vertigo, nausea, impairment in the field of vision or paresthesia were not reported.
The patient was in a good clinical condition at the latest follow-up investigation on May 2014.
Histology. The tumor exhibited chondroid differentiation, with tumor cells featuring abundant light and basophilic cytoplasm (Figure 2). The nuclei were small and sometimes polygonal. Distinct cellular atypia was rarely observed, and cell density was low. Mitotic activity was also low. The immunohistochemical analysis showed a low proliferative index (5% maximum), as highlighted by MIB1 staining. All tumor cells were strongly positive for S-100 protein, and negative for epithelial membrane antigen (EMA) and pankeratin (Figure 3).
Thus, a chondroid differentiated neoplasia, corresponding to a highly to moderately differentiated chondrosarcoma (G-1 or G-2) was diagnosed.
Discussion and Review of the Literature
According to a literature review by Bloch and colleagues, where 560 patients suffering from intracranial chondrosarcoma were analyzed, the overall 5-year mortality rate among these patients was 11.4%, and the average survival time was 53.7 months (4). The most frequent tumor location reported in their article was the clivus (32%), and the second most common location was the temporo-occipital junction (27%). Diplopia was the most common symptom at presentation (11%), and headache was the second most common symptom (9%). A total of 161 out of 560 patients were treated with surgery alone, whilst 325 patients were treated with surgery additionally to postoperative adjuvant irradiation. The 5-year mortality rate was higher in patients that were treated with surgery only (4). The histological subtype was also found to influence the prognosis, as the mortality rate was significantly lower among patients suffering from conventional chondrosarcoma compared to those suffering from the mesenchymal subtype (6% vs. 54%; p<0.0001). Not surprisingly, the histological grade was found to be associated with prognosis. The lowest mortality rate was observed among patients suffering from grade I chondrosarcoma (5%), and the highest occurred in patients suffering from grade III chondrosarcoma (25%), while patients with grade II disease had a mortality rate of 10% (4).
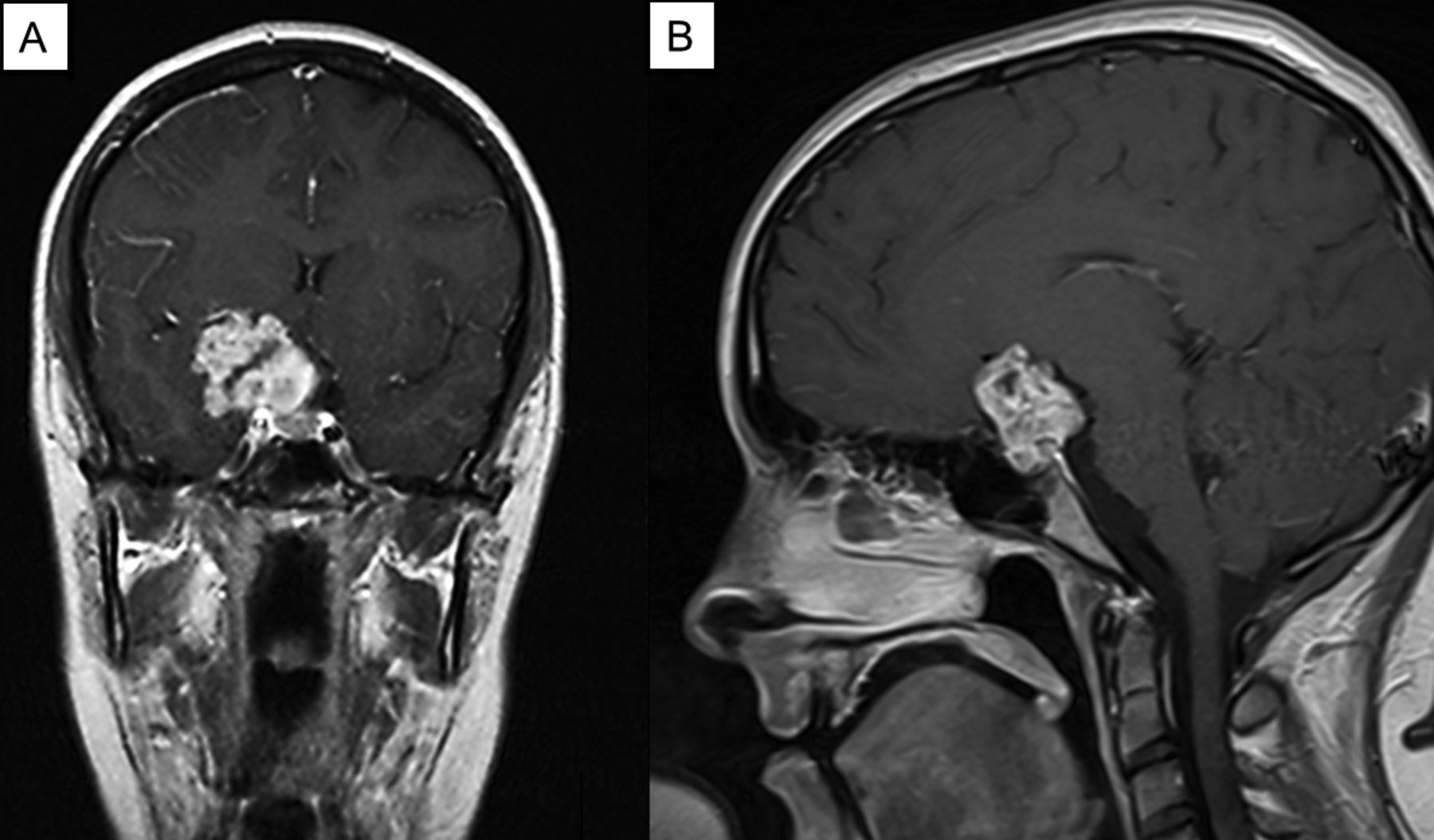
Magnetic resonance imaging of the brain, T1-weighted image. Coronal section (A), sagittal section (B).
Many of the previous case reports on intracranial chondrosarcomas suggest a particularly poor prognosis. In an analysis of primary intracranial soft tissue sarcomas in children and adolescents, a patient with intracranial chondrosarcoma was also included, and early local relapse occurred in this case (18).
Korten and colleagues outlined 15 cases of intracranial chondrosarcoma (5): nine patients were male and six were female. The tumor location was parasellar in four patients, the fossa media in three patients, around the clivus in three patients and the pontomedullary angle, os petrosum, behind the os petrosum, and at the foramen jugulare in one patient each, respectively. In one patient, the tumor location was not known. Seven out of the 15 patients were diagnosed with grade I chondrosarcoma, and six with grade II disease. None of the patients was diagnosed with grade III chondrosarcoma. Signs and symptoms included headache, diplopia, sensory disturbance or paresthesia, headache, strabism, dysdiadochokinesis and ataxia (5). Overall, 13 out of 15 patients underwent surgical resection. Postoperative proton radiotherapy was conducted in two patients, and one patient received postoperative conventional radiotherapy. Tumor recurrence was reported for seven (54%) patients. The mean time to recurrence was three years. The recurrence-free survival rate was 67% at two years, and 43% at five years (5). Korten et al. also reviewed 177 cases of intracranial chondrosarcoma concerning the clinical presentation, pathological characteristics, radiological data and treatment options (5). The mean age of the patients in this review was 37 years, and the male-to-female ratio was 1:1.1. Diplopia with impairment of eye movement was the most frequent symptom at hospital presentation (51%). The location of the chondrosarcoma was the petrosal bone in most cases (37%), the occipital bone and clivus in 23%, the sphenoid bone in 20% and the frontal, ethmoidal and parietal bones in 14% (5). In the radiographic examinations, signs of bone destruction and calcification and the involvement of neuronal and vascular structures were visible. Based on the histological analyses, 51% of the tumors were grade I lesions, 11% were grade II, 30% were of the mesenchymal type and 8% of the myxoid type (5). Surgery was conducted in nearly all of the patients. Notably, regrowth of the tumor after surgical resection occurred in 53% of the patients (after 32 months on average). Postoperative adjuvant radiotherapy was given to 20-44% of the patients (5). In a subset of patients suffering from low-grade chondrosarcoma of the skull base, combined proton and photon radiotherapy and helium or neon radiotherapy was performed (19, 20). Thereby local control rates of 78%-91%, and survival rates of 83%-94% were achieved at five years.
A primary intracerebral mesenchymal chondrosarcoma featuring rhabdomyosarcomatous differentiation was described by Marshman and colleagues (21). Noticeably, intracranial chondrosarcomas outside the brain parenchyma are much more common than intraparenchymal ones (21, 22). In their case, a 17-year-old Asian girl presented to hospital because of a 4-week history of headache, vomiting and paraesthesia in her left hand. Further clinical examination revealed incomplete left hemiparesis, a dilated right pupil and bilateral papilledema. A calcified, contrast-enhancing tumor located in the right temporo-parietal region was verified on computed tomography (CT). A midline shift was also observed on the CT image. After radical tumor excision and initial clinical recovery, local recurrence happened rapidly, before any radiotherapy was administered. The histopathological analysis revealed a mesenchymal chondrosarcoma featuring osseous and rhabdomyosarcomatous differentiation. A second radical excision was performed and unfortunately, local recurrence occurred. The recurrent tumor was considered inoperable. Within three weeks, the patient had died. This case report emphasizes the especially aggressive behavior of mesenchymal chondrosarcomas (21).

Hematoxylin and eosin stain of the intracranial chondrosarcoma. Original magnification: A, B: ×20; C, D: ×40.

{kind=link}
{kind=link}
{kind=link}
The immunohistochemical stain for epithelial membrane antigen (A) and keratin (B) reveals negative results. The MIB1 stain (C) indicates a low proliferative index, with only few cells featuring positive immunoreactivity (arrow). The S-100 stain (D) is strongly positive. Original magnification, ×40.
Wu et al. outlined two cases of primary non-skeletal intracranial cartilaginous neoplasms in 1970 (23). Neither the chondroma nor the mesenchymal chondrosarcoma primarily originated from the skull bone but were located intracerebrally, with dura mater separating the tumors from the skull bone. The chondroma was surgically resected, and no tumor recurrence was reported. The mesenchymal chondrosarcoma was also resected, but local recurrence occurred rapidly. In spite of cobalt therapy and numerous secondary resections, death occurred 16 months after the initial surgery (23).
Another mesenchymal intracranial chondrosarcoma was reported by Kan and colleages (24). A 31-year-old woman presented to hospital with strong headache, nausea and vomiting for one week. The clinical investigation revealed no conspicuity. On MRI, however, a giant heterogeneous and intensely enhancing tumor mass of 6×5×4 cm was seen. The tumor was located around the anterior falx cerebri. The patient underwent extensive tumor excision, and in the histopathological analysis, a mesenchymal chondrosarcoma was diagnosed (24). The authors emphasize that a combination of microsurgical resection and radiotherapy should be used as therapy of choice for intracranial chondrosarcomas (24).
Khan et al. have outlined a case of a sphenoidal intracranial chondrosarcoma (25). A 40-year-old man with a clinical history of headache and decreased vision of the left eye, and complete loss of vision of the right eye presented to hospital. In the fundoscopy, secondary optic atrophy in the right eye and temporal paleness of the optic disc of the left eye were seen. The MRI of the brain showed a tumor mass arising from the sphenoid sinus, eroding the overlying bone, with a spreading into the sellar and parasellar region. The tumor mass was compressing the optic nerve. After excisional biopsy, the histological examination revealed a chondrosarcoma. The patient underwent extensive surgical resection followed by proton radiation therapy (25). The outcome is not known, since this case was described just recently.
Summarizing these data, intracranial chondrosarcomas have a poor prognosis with a high chance of local recurrence (4, 5). Not surprisingly, surgery alone seems to be a less effective treatment strategy compared to surgery combined with adjuvant radiotherapy (4). Thereby, the best long-term outcomes are achieved (26, 27). In 1999 Rosenberg et al. conducted the largest single-Center study on chondrosarcomas of the skull base (28). In this study, the whole treatment was carried out at one single institution and 200 patients were enrolled. When surgery was combined with irradiation, the 5- and 10-year survival rates were 99% in this study (28). It remains to be investigated whether surgery and adjuvant radiotherapy is also the best treatment option for low-grade intracranial chondrosarcomas. Possibly, surgery-alone or delayed adjuvant radiation therapy is a better option in low-grade cases, keeping in mind the well-reported side-effects of cranial irradiation.
Conclusion
Intracranial chondrosarcomas should be considered as differential diagnosis for intracranial tumors, especially when located at the skull base. Therapy should imply extensive surgical excision, followed by radiotherapy. Neoadjuvant irradiation may reduce tumor size, when the tumor is primarily considered unresectable. Previously published data about intracranial chondrosarcomas indicate the extreme aggressiveness of this tumor entity.
Footnotes
-
Conflicts of Interest
The Authors declare that they have no affiliations with or involvement in any organization or entity with any financial interest, or non-financial interest (such as personal or professional relationships, affiliations, knowledge or beliefs) in the subject matter or materials discussed in this article.
- Received October 13, 2014.
- Revision received November 2, 2014.
- Accepted November 5, 2014.
- Copyright© 2015 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved