

Abstract
Background: Zoledronic acid (ZOL) used for the prevention/treatment of osteopathic complications has been reported to have antitumor effects in breast cancer treatment. However, little is known about the exact molecular mechanisms for antitumor actions of ZOL. In this study, two breast cancer cell lines were used to investigate the antitumor efficacy of ZOL and the underlying molecular mechanisms. Results: The growth of two breast cancer cell lines was markedly decreased following treatment with ZOL. Compared with MCF-7 cells, MDA-MB-231 cells were more sensitive to ZOL treatment. Western blot analysis showed that the inhibitory effect of zoledronic acid on growth was related to the extent of inhibition of phosphorylated-protein kinase B (p-AKT), and phosphorylated-mammalian target of rapamycin (p-mTOR). Moreover, the expression of the stress-responsive protein regulated in development and DNA damage response 1 (REDD1), an inhibitor of mTOR, was induced markedly to various degrees in different breast cancer cell lines after ZOL treatment. Interestingly, by examining the upstream signaling pathway of REDD1, we found that ZOL can induce endoplasmic reticulum stress responses through activating the protein kinase R (PKR)-related ER kinase-eukaryotic initiation factor 2 alpha-CCAAT/enhancer binding protein homologous protein (PERK-eIF2α-CHOP) pathway. Conclusion: Taken together, these results indicated that ZOL-induced cell death was caused by endoplasmic reticulum stress activating PERK-eIF2α-CHOP pathway to induce REDD1 expression and inhibit the mTOR pathway.
- Breast cancer
- zoledronic acid
- mTOR
- REDD1
- endoplasmic reticulum stress
- MCF-7
- MDA-MB-231 cells
Breast cancer remains the second and fourth leading cause of cancer in women in America and Taiwan, respectively, despite rapid treatment advances in recent years. Often associated with breast cancer is high frequency of tumor metastasis to other organs, including the lungs, liver or bone, which is the major cause of death for patients with breast cancer (1). In bone metastasis, osteoclast-mediated bone resorption is stimulated by metastatic cells, resulting in osteolysis, bone fragility and proneness to fracturing (2). Bisphosphonates, such as zoledronic acid (ZOL), are potent inhibitors of osteoclast-mediated bone resorption (3-6) and are therefore widely used in the treatment of bone metastasis and bone diseases with a high bone turnover, such as Paget's disease (7), tumor-associated osteolysis (8), and post-menopausal osteoporosis (9). Although the main target of bisphosphonates is thought to be the osteoclast, there is increasing evidence to indicate that bisphosphonates have direct antitumor activity (3, 10). Indeed, in vitro studies have shown that ZOL is the bisphosphonate with the most potent antitumor effects, including induction of tumor cell apoptosis and inhibition of tumor cell proliferation (11-13). ZOL has also been reported to have antiangiogenesis and anti-invasion effects in breast cancer (14, 15). Combination of ZOL with other chemotherapy drugs for the treatment of breast or prostate cancer has shown increased antitumor efficacy than single-agent therapy (16-18). However, the exact biochemical mechanisms for antitumor action of ZOL are still poorly understood.
It has been reported that ZOL inhibits growth and survival of pancreatic cancer cells by functional down-regulation of the mitogen-activated protein kinases (MAPK) and phosphoinositol 3-kinase/protein kinase B (PI3K/AKT) pathways (19). Mammalian target of rapamycin (mTOR), associated with tumorigenesis, is one important downstream effector of the PI3K/AKT signaling pathway (20). Therefore, inhibition of the mTOR signaling pathway might be a possible mechanism of ZOL-induced growth inhibition. On the other hand, regulated in development and DNA damage response 1 (REDD1) is another key factor inhibiting the phosphorylation of mTOR (21, 22), resulting in protein synthesis inhibition and tumor suppression (23). REDD1, identified as a cell stress-responsive protein, can be induced by stress signals including hypoxia, endoplasmic reticulum (ER) stress and reactive oxygen species (ROS) that cause cell apoptosis and DNA damage (22, 24). Therefore, REDD1 might be a candidate molecule involved in ZOL-induced antitumor effects.
It has been shown that ZOL inhibits farnesyl pyrophosphate synthase (FPPS) activity in the mevalonate pathway to induce ER stress (25). The ER is an organelle for membrane protein synthesis, initial post-translational modification, folding and secretion (26). When cells are stimulated by nutrient or oxygen deficiency, homeostasis of the ER is disrupted, producing a stress known as ER stress, which then initiates a host of stress reactions, the general directions of which are stress amelioration and restoration of homeostasis (27). Among these is the stimulation of chaperone proteins which in turn triggers multiple protective cellular responses to reduce unfolded protein, a process known as the unfolded protein response (UPR) (28). However, the presence of excessive unfolded proteins induces caspases to trigger apoptosis and results in cell death (29). There are three ER proximal sensors to regulate UPR, including protein kinase R (PKR)-related ER kinase (PERK), inositol-requiring enzyme 1 (IRE1) and activating transcription factor 6 (ATF6). PERK is a serine threonine kinase. Under stress, PERK promotes eukaryotic initiation factor 2 alpha (eIF2α) phosphorylation to reduce protein translation and induce CCAAT/enhancer binding protein homologous protein (CHOP) to lead to cell apoptosis (30). Moreover, previous studies indicated that ER stress increased REDD1 expression in many cell types (31, 32). In eIF2α kinase PERK-deficient mouse embryonic fibroblasts, ER stress cannot promote REDD1 expression, suggesting that eIF2α phosphorylation is necessary for the induction of REDD1 in the ER stress response.
In this study, the direct effect of ZOL on the growth and survival of metastatic breast cancer cells were examined. The molecular mechanism of ZOL-induced differential growth inhibition in breast cancer cells was investigated. The AKT/mTOR signaling pathway, REDD1 expression and ER stress response were evaluated after ZOL treatment.
Materials and Methods
Cell culture. The human breast cancer cell lines MCF-7 and MDA-MB-231 obtained from American Type Culture Collection (ATCC, Manassas, VA, USA) were cultured in Dulbecco's modified Eagle's medium (DMEM; Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS; Hyclone, Logan, UT, USA), 1% L-glutamine (Invitrogen), 1% non-essential amino acid (Invitrogen) and 1% penicillin-streptomycin-amphotericin B (Biological Industries, Kibbutz beit Haemek, Israel) at 37°C in a humidified atmosphere containing 5% CO2.
Cytotoxicity assay (MTT assay). MTT assay was performed to determine the effect of ZOL on cell growth of breast cancer cells. Briefly, cells were seeded in a 96-well culture plate at a density of 7×103 cells/ml and then treated with 0.1, 1, 10, 50, or 100 μM ZOL for 24, 48 and 72 h. After drug treatment, 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide (MTT; Sigma, St Louis, MO, USA) was added and cells were incubated for two hours at 37°C in the dark. The resulting formazan product was dissolved in dimethyl sulfoxide (DMSO; Sigma) and optical density (OD) values were measured using a microplate reader (SpectraMax 250; Molecular Devices, Sunnyvale, CA, USA) at an absorption wavelength of 570 nm. Data are expressed as the fraction of cells inhibited by drug treatments compared with vehicle-treated cells.
Measurement of apoptosis. To analyze cellular DNA contents, ZOL-treated cells were collected, fixed in 70% ice-cold ethanol, and kept at −20°C for at least 14 h. The fixed cells were washed twice with phosphate buffered saline (PBS), stained with propidium iodide (PI; 50 μg/ml) in PBS containing 0.2% Tween 20 and 2.5 μg/ml RNase at 4°C overnight, and analyzed by flow cytometry. The profiles of cellular DNA contents indicated the distribution of the cells in different phases of the cell cycle. The cells with DNA content less than that of the G0/G1 cells were classified as the sub-G0/G1 population and considered as apoptotic cells.
Western blot analysis. Cells were scraped away from 10-cm dishes and suspended in RIPA lysis buffer (50 mM Tris-HCl, 150 mM NaCl, 0.25% sodium deoxycholate, 1% Triton X-100, pH 7.4) on ice. After centrifugation (13,000 ×g) at 4°C for 15 min, the protein concentration was determined by Bio-Rad protein assay (Bio-Rad, Hercules, CA, USA). Protein lysates (30 μg) were separated in 8-15% sodium dodecyl sulfate (SDS)-polyacrylamide gels and then transferred to a nitrocellulose membrane. The membrane was blocked with 5% milk in 1× TBST buffer (10 mM Tri-base, 150 mM NaCl, 0.05% Tween 20, pH 7.4) for one hour at room temperature. Primary antibodies including those against REDD1 (Proteintech, Chicago, IL, USA), mTOR, p-mTOR, PERK, IRE1α, CHOP (Cell Signaling, Danvers, MA, USA), ATF6 (Abcam, Cambridge, MA, USA), p-PERK (Santa Cruz Biotechnology, Santa Cruz, CA, USA), eIF2α, p-eIF2α (Invitrogen) and β-actin (Sigma) were then used to probe these blots at 4°C overnight. Membranes were washed with TBST three times and then incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies for two hours. Following TBST washing, protein visualization was carried out using an enhanced chemiluminescence kit (ECL; Millipore, Bedford, MA, USA).
Reverse transcription and real-time quantitative polymerase chain reaction (qPCR). Total RNA was extracted and precipitated according to the instructions of the manufacturer (TRIzol® Reagent; Invitrogen). RNA was dissolved in diethylpyrocarbonate (DEPC)-treated water (Invitrogen) and denatured at 65°C for 10 min. After denaturation, the sample was put on ice immediately for one minute. For reverse transcription, RNA was incubated with ready-to-go you-prime first-strand beads (GE Healthcare Biosciences, Pittsburgh, PA, USA) and 2.5 μM random primer dN6 at 37°C for one hour. cDNA samples were then mixed with forward and reverse primer (REDD1: forward 5’ GTT TGA CCT CTC CAC CAG CCT 3’, reverse 5’ GCA CAC AAG TGT TCA TCC TCA GG 3’; β-actin: forward 5’ TGG CAT TGC CGA CAG GAT 3’, reverse 5’ GCT CAG CAG CAG CAA TGA TCT 3’), 2× Fast SYBR PCR Master Mix (Applied Biosystems, Carlsbad, CA, USA) and 6 μl ddH2O. The PCR reaction was carried out with an initial denaturation at 95°C for 10 minutes, followed by 50 cycles at 95°C for 3 s and 60°C for 30 s for real-time fluorescence quantitation of products.
Statistical analysis. The means and standard deviation (SD) were calculated from three independent experiments. Two-way ANOVA with post-hoc test was used for multi-group comparisons. Differences were considered statistically significant when p<0.05.
Results
The effect of zoledronic acid on cell growth. The MTT assay was used to examine the anti-proliferation effect and determine the inhibitory dose of ZOL in MCF-7 and MDA-MB-231 breast cancer cells. Comparing to untreated controls, growth of MCF-7 and MDA-MB-231 cells significantly decreased when treated with 50 and 100 μM ZOL for 48 and 72 h (Figure 1). The results also showed that the growth inhibition of ZOL-treated MCF-7 cells was cytostatic. However, the growth inhibition of ZOL-treated MDA-MB-231 cells occurred in a time- and dose-dependent manner, indicating that ZOL induced a cytotoxic effect on MDA-MB-231 cells.
To further evaluate the antiproliferative effect of ZOL, we checked the cell-cycle phase distribution of ZOL-treated breast cancer cells by flow cytometry (Figure 2). The results showed that treatment of MCF-7 cells with 50 μM of ZOL resulted in S-phase accumulation and an increased proportion of sub-G0/G1 cells (9.34%) in a time-dependent manner (p<0.001, Figure 2A and C). However, compared to 0 or 10 μM treatment, treatment with 50 μM of ZOL significantly increased the proportion of sub-G0/G1 MDA-MB-231 cells (31.46%) in a time-dependent manner (p<0.001, Figure 2B and D). These results together suggest that ZOL induced more significant apoptosis in MDA-MB-231 cells than in MCF-7 cells.
The effect of zoledronic acid on the AKT/mTOR signaling pathway. Since ZOL demonstrated an antiproliferative activity towards breast cancer cells, we next examined the underlying growth inhibitory effect of ZOL. We firstly evaluated the expression of AKT/mTOR signaling pathway proteins by western blot analysis. After treatment with 50 μM ZOL for 24, 48 and 72 h, the expression of phosphorylated-AKT (p-AKT), AKT, phosphorylated-mTOR (p-mTOR) and mTOR in the MCF-7 cells were similar to those of the untreated controls (Figure 3A). However, in contrast to MCF-7 cells, the expressions of p-AKT, p-mTOR and mTOR in ZOL-treated MDA-MB-231 cells were obviously inhibited as compared to those of the untreated controls (Figure 3). The results together suggest that inhibition of p-AKT, and p-mTOR by ZOL related well to the ZOL-induced cytotoxic effects.

The effects of zoledronic acid (ZOL) on the growth of breast cancer cells. MCF-7 (A) and MDA-MB-231 (B) cells were treated with ZOL at the indicated concentrations for 24, 48 and 72 h and cell growth was determined by the MTT assay. Data are expressed as the mean±S.D. of three replicates and are representative of three independent experiments. *Significantly different as compared to the control at the same time point (p<0.001).

The effects of zoledronic acid (ZOL) on the cell cycle of breast cancer cells. MCF-7 (A) and MDA-MB-231 (B) cells were treated with ZOL at the indicated concentrations for 24, 48 and 72 h and then the cell cycle distribution was determined by flow cytometry. The percentage of apoptotic sub-G0/G1 cells from three independent experiments are expressed as the mean±S.D. and shown for MCF-7 (C) and MDA-MB-231 (D) cells. *Significantly different as compared to the control at the same time point (p<0.001). Apop: apoptotic sub-G0/G1 cells.
The effect of zoledronic acid on REDD1 expression. In addition to p-AKT, REDD1 is another key factor that regulates cell growth through modulating the mTOR signaling pathway. Thus, we examined the mRNA level and protein expression of REDD1 in ZOL-treated breast cancer cells by real-time qPCR and western blot analysis. The mRNA levels of REDD1 in MCF-7 and MDA-MB-231 cells were significantly increased after 50 μM ZOL treatment for 24 and 48 h (Figure 4A and B). The protein expression of REDD1 in ZOL-treated MCF-7 and MDA-MB-231 cells was also significantly increased as compared to that in controls (Figure 4C). After treatment with ZOL for 24 h, the fold of increase of REDD1 was 1.1 and 1.8 for MCF-7 and MDA-MB-231 cells, respectively. At 48 h after ZOL treatment, the fold of increase of REDD1 was 1.3 and 2.7 for MCF-7 and MDA-MB-231 cells, respectively. Interestingly, the fold of increase of ZOL-induced REDD1 mRNA and protein were higher in the MDA-MB-231 cells as compared to the MCF-7 cells. These results together showed that REDD1 is involved in the molecular mechanism of ZOL-induced cytotoxicity and ZOL inhibited mTOR signaling pathway through inducing REDD1 expression.
The effect of zoledronic acid on ER stress responses. It has been reported that REDD1 can be up-regulated by the ER stress response. Thus, ER stress response-related proteins including IRE1α, ATF6, PERK, eIF2α and CHOP were evaluated in breast cancer cells after treatment with 50 μM ZOL for 24 and 48 h. Comparing to those of the controls, the expressions of ATF6, PERK and CHOP were significantly up-regulated in MDA-MB-231 cells treated with ZOL for 24 and 48 hours (Figure 5). ZOL also activated the expression of phosphorylated-PERK and -eIF2α (p-PERK and p-eIF2α), but had no effect on the expression of IRE1α and eIF2α. These results reveal that ZOL treatment can induce ER stress responses through activating PERK-eIF2α-CHOP and ATF6 signaling pathways in MDA-MB-231 cells.

The effects of zoledronic acid (ZOL) on protein kinase B/mammalian target of rapamycin (AKT/mTOR) signaling pathways of breast cancer cells. MCF-7 (A) and MDA-MB-231 (B) cells were treated with 50 μM ZOL for 24, 48 and 72 h. Western blot analysis was used to detect the expression of p-AKT, AKT, p-mTOR and mTOR in breast cancer cells. β-Actin was used as a loading control.
Discussion
In this study, our results clearly showed that the efficacy of ZOL was higher in MDA-MB-231 cells than MCF-7 cells, including cell growth inhibition and apoptosis induction. We also found a new pathway of ZOL-induced growth inhibition. ZOL induced ER stress to activate the PERK-eIF2α-CHOP pathway followed by increasing REDD1 expression, then inhibited the mTOR signaling pathway to result in cell growth inhibition.
We found that MCF-7 and MDA-MB-231 cells treated with ZOL exhibited significant accumulation of cells in the S phase (Figure 2). However, significantly more sub-G0/G1 cells were observed for MDA-MB-231 cells than MCF-7 cells. These results suggest that S phase accumulation is not a prerequisite for the increase of sub-G0/G1 cells. These findings suggest that ZOL-induced cell growth inhibition occurs not only through inhibiting the cell cycle, but also by affecting other intracellular signaling pathways.
It is interesting that the mechanism of cell growth inhibition by ZOL differs in different types of breast cancer cells. Previous studies showed that ZOL can down-regulate PI3K/AKT and MAPK pathways to inhibit growth or induce apoptosis in prostate gland and pancreatic cancer cells (19, 33). Indeed, AKT can directly and indirectly regulate inhibitor of nuclear factor kappa-B kinase (IKK) activity to cause nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) to move into the nucleus, and activation and anti-apoptosis gene transcription (34). AKT can promote B-cell lymphoma 2 (BCL2) family expression to inhibit cell apoptosis and increase cell survival (35). Therefore, inhibition of AKT activity has usually been associated with antiproliferative effects or apoptotic responses. These results corresponded well with our findings that ZOL-induced cytotoxicity may be mediated through inhibiting p-AKT expression in MDA-MB-231 cells but not in MCF-7 cells (Figure 3). Moreover, significant inhibition of p-mTOR expression was observed in ZOL-treated MDA-MB-231 cells but not in MCF-7 cells (Figure 3), which also explains why ZOL had different inhibitory effects in these two breast cancer cell lines. Together, these results suggest that ZOL inhibited growth and induced cell apoptosis in breast cancer cells through inhibiting the AKT/mTOR signaling pathway.
It has been reported that REDD1 can promote tuberous sclerosis complex 1/2 (TSC1/2) activation and mTOR Complex 1 (mTORC1) inhibition to cause tumor suppression (23). A previous study also reported that REDD1 promoted the apoptotic death of differentiated neuronal PC12 cells and led to their sensitization to hypoxic and oxidative stress (36). In this study, we observed that ZOL enhanced the expression of REDD1 at both mRNA and protein levels in MCF-7 and MDA-MB-231 cells (Figure 4). Compared to MCF-7 cells, MDA-MB-231 cells are more sensitive to ZOL and showed higher level of ZOL-induced REDD1. These results together suggest that REDD1 may be centrally involved in the drug sensitivity of breast cancer cells to ZOL.
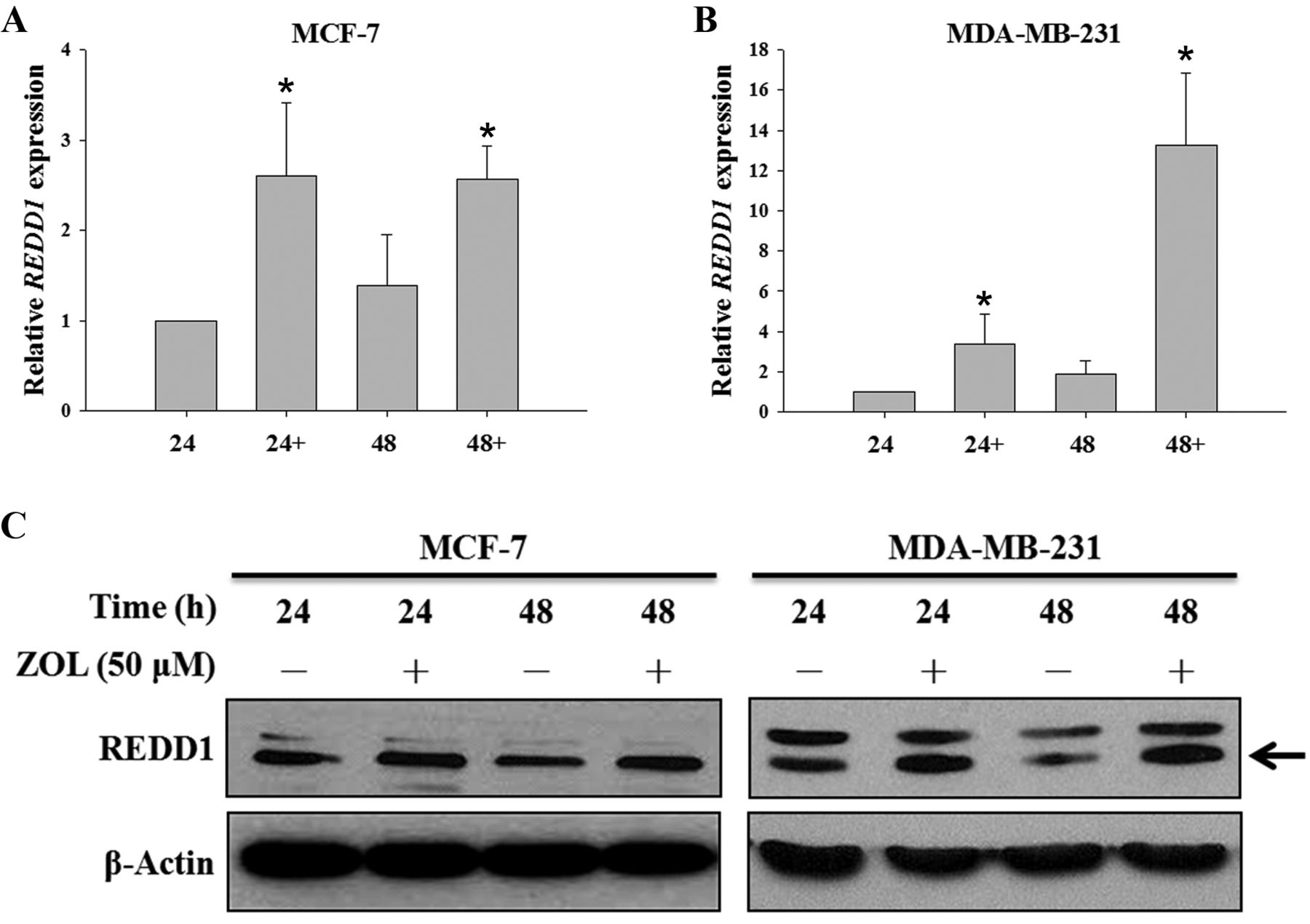
The effects of zoledronic acid (ZOL) on the regulated in development and DNA damage response 1 (REDD1) mRNA and protein expression of breast cancer cells. MCF-7 (A) and MDA-MB-231 (B) cells were treated with 50 μM ZOL (+) for 24 and 48 hours and relative mRNA expression was determined by quantitative polymerase chain reaction. Western blot analysis was used to detect the expression of REDD1 (arrow) in breast cancer cells (C). β-Actin was used as a loading control.
Our results show that MDA-MB-231 cells but not MCF-7 cells were more sensitive to ZOL through inducing REDD1 and inhibiting mTOR signaling pathway (Figures 3 and 4). However, a different mechanism of action of ZOL on breast cancer cells has also been suggested. Espinoza et al. found that differently from non-cysteine-rich protein 61 (CCN1)-expressing MCF-7 cells, MDA-MB-231 cells have high CCN1 expression and are more sensitive to ZOL by down-regulation of CCN1 expression in a dose-dependent manner (37). Indeed, CCN1 a survival factor, is overexpressed in 30% of patients with metastatic breast cancer has the ability to induce tumorigenesis and angiogenesis, thus down-regulation of CCN1 expression by ZOL could lead to cell growth inhibition and apoptosis (37, 38). Briefly, it is worth further investigating the relation between CCN1, REDD1 and mTOR pathways in ZOL-induced cell death.
ZOL has been reported to inhibit FPPS in the mevalonate pathway and then induce ER stress (25). ER stress was found to trigger the PERK/eIF2α/ATF4 pathway and up-regulate REDD1 expression (32, 39). This suggests that ZOL may induce REDD1 expression through activation of the ER stress response. Indeed, our results showed that p-PERK and p-eIF2α were activated and REDD1 expression was increased simultaneously in breast cancer cells on ZOL treatment (Figures 4 and 5). Moreover, in a preliminary study using eIF2α-mutated breast cancer cells, we observed that abolishment of the ER stress response attenuated the inhibitory effect of ZOL by reducing REDD1 expression and increasing mTOR activation (data not shown). Therefore, these results together suggest that ZOL-induced cell death was mediated by ER stress activating the PERK-eIF2α-CHOP pathway to induce REDD1 and inhibit the mTOR signaling pathway.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The effects of zoledronic acid (ZOL) on endoplasmic reticulum (ER) stress pathway of MDA-MB-231 cells. Cells were treated with 50 μM ZOL for 24 and 48 hours. Western blot analysis was used to detect the expression of inositol-requiring enzyme 1 alpha (IRE1α), activating transcription factor 6 (ATF6), phosphorylated-protein kinase R (PKR)-related ER kinase (p-PERK), PERK, phosphorylated-eukaryotic initiation factor 2 alpha (p-eIF2α), eIF2α, and CCAAT/enhancer binding protein homologous protein (CHOP) in MDA-MB-231 cells. β-Actin was used as a loading control.
Recently, several clinical trials have emphasized on the antitumor activity of ZOL. Combinational treatment of letrozole and ZOL improved the disease-free survival and recurrence rate as compared with letrozole alone (40). Additionally, chemotherapy combined with ZOL may improve disease-free survival and overall survival of breast cancer patients with estrogen receptor-negative tumors (41). Thus, ZOL is a potential drug for killing breast cancer, and our findings that ZOL modulates the REDD1-mTOR signaling pathway will provide important clues for future application in breast cancer therapy.
Acknowledgements
We thank Novartis for providing zoledronic acid. This work was supported by grant V100C-119 from the Taipei Veterans General Hospital, Taiwan.
Footnotes
-
* These Authors contributed equally to this work.
- Received June 20, 2013.
- Revision received July 13, 2013.
- Accepted July 16, 2013.
- Copyright© 2013 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved