

Abstract
Background: Candidate of metastasis-1 (COM-1) is a molecule which is stress-induced and cell growth-related. The current study investigated the impact of COM-1 and role of Peroxisome Proliferating Activator Receptor-gamma (PPARγ) agonists on COM-1 knock down in colorectal cancer cells. Materials and Methods: Expression of COM-1 was knocked out in the colorectal cancer cell lines RKO and CaCO2 by transfection with ribozyme transgenes which specifically targeted COM-1. Cell growth, cell migration and apoptosis were measured in wild-type, control and COM-1 knock down cells. The impact or treatment with the PPARγ agonists on the function of COM-1 knock down cells was monitored. Results: COM-1 knock-down cells grew slower than wild-type and control cells. When treated with the PPARγ agonist ciglitazone and bisphenol A diglycidyl ether (BADGE) at several different concentrations, all types of cells grew slower to some extent. Cells with COM-1 knock-down had the same mobility as wild-type and control cells. More apoptotic and dead cells were detected when cells were treated with ciglitazone at 3 μM, especially of COM-1 knock down cells. Conclusion: COM-1 is an anti-apoptotic gene and a cell growth promoter. Furthermore, the PPARγ agonist could increase the inhibitory effect seen in COM-1 knock-down cell growth and to promote apoptosis. In regards to metastasis, COM-1 appears to play no role in cell motility.
- COM-1
- p8
- NUPR1
- colorectal cancer; cell growth
- cell migration
- apoptosis
- PPARγ agonist
Candidate of metastasis-1 (COM-1, also known as nuclear protein 1) was identified originally from isolated micro-metastases in the bone marrow of a patient with breast cancer and was found to be significantly up-regulated in metastases formed in the central nervous system (1). In a later paper, COM-1 was found to be identical to a protein known as p8 which had been discovered two years earlier, and which was found to be strongly activated in pancreatic acinar cells during the acute phase of pancreatitis and acted as a promoter of cellular growth factor (2). Moreover, COM-1also has a role in processing cell growth and apoptosis.
The function of COM-1 appears to be complex; it differs in various types of cancers and is even contradictory between in vivo and in vitro work in the same malignancies. In some types of cancers the expression of COM-1 in tumour cells is reduced, such as in prostate cancer (3). The knock down of COM-1 has been shown to enhance the invasiveness and growth of prostate cancer cells, thus it was concluded that COM-1 acts as a putative tumour suppressor in prostate cancer (3). In contrast, COM-1 is overexpressed in pancreatic cancer (4). It has also been suggested that COM-1 overexpression may be related to the progression and metastasis of thyroid neoplasm (5). In prostate cancer cells, it COM-1 was found to be cross-precipitated with a coactivator of peroxisome proliferator activated receptor (PPAR)γ, PGC-1, but not with PPARγ itself (3). Elimination of COM-1 from prostate cancer cells resulted in a reduced response of the cells to ciglitazone, a PPAR-gamma agonist that can inhibit growth, whereas forced expression of COM-1 rendered cells more responsive to ciglitazone (3).
There has been considerable interest in COM-1 and its expression in many tumour types, although so far, little work has been carried out on colorectal cancer, despite it being the third most commonly diagnosed tumour in the world, especially in developed countries. Davies et al. (6) described COM-1 as a tumour suppressor in human colorectal carcinoma tissues. To our knowledge, there have been no studies to determine the role of COM-1 in colorectal cancer cell lines in vitro. The aim of this study therefore, was to investigate the possible effect of COM-1 in colorectal cancer cell lines.
PCR primer sequences used in this study
Materials and Methods
Cell lines. The cell lines used were the human colorectal cancer cell lines, RKO, CaCO2 and HRT-18, which were purchased from the European Collection of Cell Culture (ECACC, Porton Down, UK). Cells were routinely cultured with Dubecco's modified Eagle's medium (DMEM, Sigma-Aldrich Company Ltd, Dorset, UK) supplemented with 10% Foetal calf serum (FCS, Sigma-Aldrich Company Ltd, Dorset, UK), penicillin and streptomycin (GIBO BRL, Paisley, UK). RNA extraction kit and reverse transcription (RT) kit were obtained from AbGene (Surrey, UK). PCR primers were designed by using Beacon Designer (Palo Alto, CA, USA) and synthesized by Invitrogen (Life Technologies, Paisley, UK). Molecular biology grade agarose and DNA ladder were obtained from Invitrogen. Master mix for routine Polymerase Chain Reaction (PCR) and quantitative PCR were from AbGene. Ciglitazone and BADGE were obtained from Sigma-Aldrich Ltd.
RNA extraction and cDNA synthesis and preparation of COM-1/p8 knockdown cells. RNA was extracted from cells using an RNA extraction kit (AbGene). The concentration of RNA was determined using a UV spectrophotometer. Reverse transcription was performed using an RT kit with an anchored oligo-dt primer (AbGene), using 0.5 μg of total RNA. The quality of the cDNA was verified by using β-actin primers. Construction of hammerhead ribozyme transgenes targeting human COM-1/p8 and COM-1 expression cassette. Hammerhead ribozymes that specifically target a GTC and a UUC site of human COM-1 (GenBank Accession AF135266, position 189 and 68, respectively), based on the secondary structure of COM-1 using Zuker's mfold programme were constructed. Touch-down PCR was used to generate the ribozyme with the primers listed in Table I. These were subsequently cloned into a pEF6/V5-His vector, using ampicillin and blasticidin as selection markers for prokaryotic and mammalian cells, respectively, and then amplified in Escherichia coli, purified, verified and used for electroporation of colorectal cancer cells. Full-length human COM-1 cDNA was generated from normal mammary tissue using primers, COM1ExF and COM1ExR (Table I). The sequence verified product was then TA cloned into pEF/V5-His and prepared in a similar manner to the ribozyme transgenes and used for transfection. The above procedure generated the following stably transfected cells from RKO and CaCO2: COM-1 knock down cells, RKOΔCOM1, CaCO2ΔCOM1; plasmid only control cells, RKOpEF and CaCO2pEF, used together with the respective wild-type cells.
In vitro cell growth assay. Wild-type cells RKO and CaCO2, control cells and COM-1 knock down cells were seeded into a 96-well plate (Nunc, Fisher Scientific, Leicester, UK) with 2000 or 5000 cells respectively in 200 μl normal medium. After three days of incubation, cells were fixed in 4% formaldehyde in buffered saline solution (BSS) for 30 minutes before being stained for 10 min with 0.5% (w/v) crystal violet. After air drying, 200 μl of 10% (v/v) acetic acid was added to each well and the absorbance was measured at a wavelength of 540 nm on a plate reading spectrophotometer (EL×800, Bio-Tek, Wolf Laboratories, York, UK). Cell growth is presented as the increase absorbance for each time point.
Motility assay by electric cell impedence sensing (ECIS). ECIS (Applied Biophysics Inc, NJ, USA) was used for motility assay (wounding assay) andwounding/cell modelling analysis in the study model. The ECIS instrument measures the impedance and capacitance of cells attached to a gold electrode. Cell modelling was carried out using the ECIS RbA modelling software, supplied by the manufacturer. The array surface was treated with 200 μl of 10 mM L-cysteine solution for 20 min, which binds to the gold surface via its thiol group forming a monomolecular layer, followed by two washes in DMEM with 15 mM Hepes and L-glutamine (Lonza Laboratories, Verviers, Belgium). An electrode check was run to check the impedance of the cell-free wells containing fresh medium alone and to assess the integrity of the arrays. The arrays were seeded at a density of 80,000 cells in 400 μl of DMEM with 15 mM Hepes, L-glutamine and the cells left to grow to achieve confluent monolayers before treating with inhibitors. The monolayer of cells was electrically wounded with a 5 V AC at 4,000 Hz for 30 s. Impedance and resistance of the cell layer were immediately recorded every millisecond for as long as required.
Cell migration assay. The migration of the cells across a wounded surface of a confluent monolayer was examined to assess the motility of cell lines. Cells at a density of 40,000 cells per well were seeded in a 24-well plate; upon reaching confluence, the medium was changed and the monolayer was scraped with a fine gauge needle to create a wound. A few drops of mineral oil were added to avoid evaporation of the medium. The plate was placed on a heated stage (Leica GmbH, Bristol, UK) to maintain a constant temperature of 37°C. Cells were photographed after wounding and every 15 min during 2 h with a CCD camera attached to a Leica DM IRB microscope (Leica GmbH, Bristol, UK) at ×20 magnification. Cell migration was analysed using Image J software (NIH, Bethesda, MA, USA) by measuring the distance between the two wounded fronts at 10 points per incubation; the arbitrary values obtained were converted into micrometers by multiplying each value by 1.6 as previously calibrated using a calibrated grid. The distance between the migrating fronts at each point time was determined by subtracting the distance between the two fronts at any of the specific times selected from that at the initial starting point (0 minutes). Wild-type cells, plasmid control cells and COM-1 knock-down cells were then seeded in the 96 well plate with 2000 cells and 5000 cells in each well respectively in 100 μl of normal medium. Before seeding cells, different concentrations of ciglitazone solution were made up by 1 in 5 fold dilution, with an initial concentration of 200μM (125 μl) ciglitazone solution in the first row of a 96-well plate, then 100 μl normal medium were added into each well from the second row to the eighth row, then 25 μl ciglitazone solution was taken from the first row to the next row until the 25 μl of the eighth row was discarded. In this way, there were 100 μl of ciglitazone solution in each well and the concentration was from 200 μM to 0.00256 μM, thus reducing the concentration of ciglitazone by half in each cell.
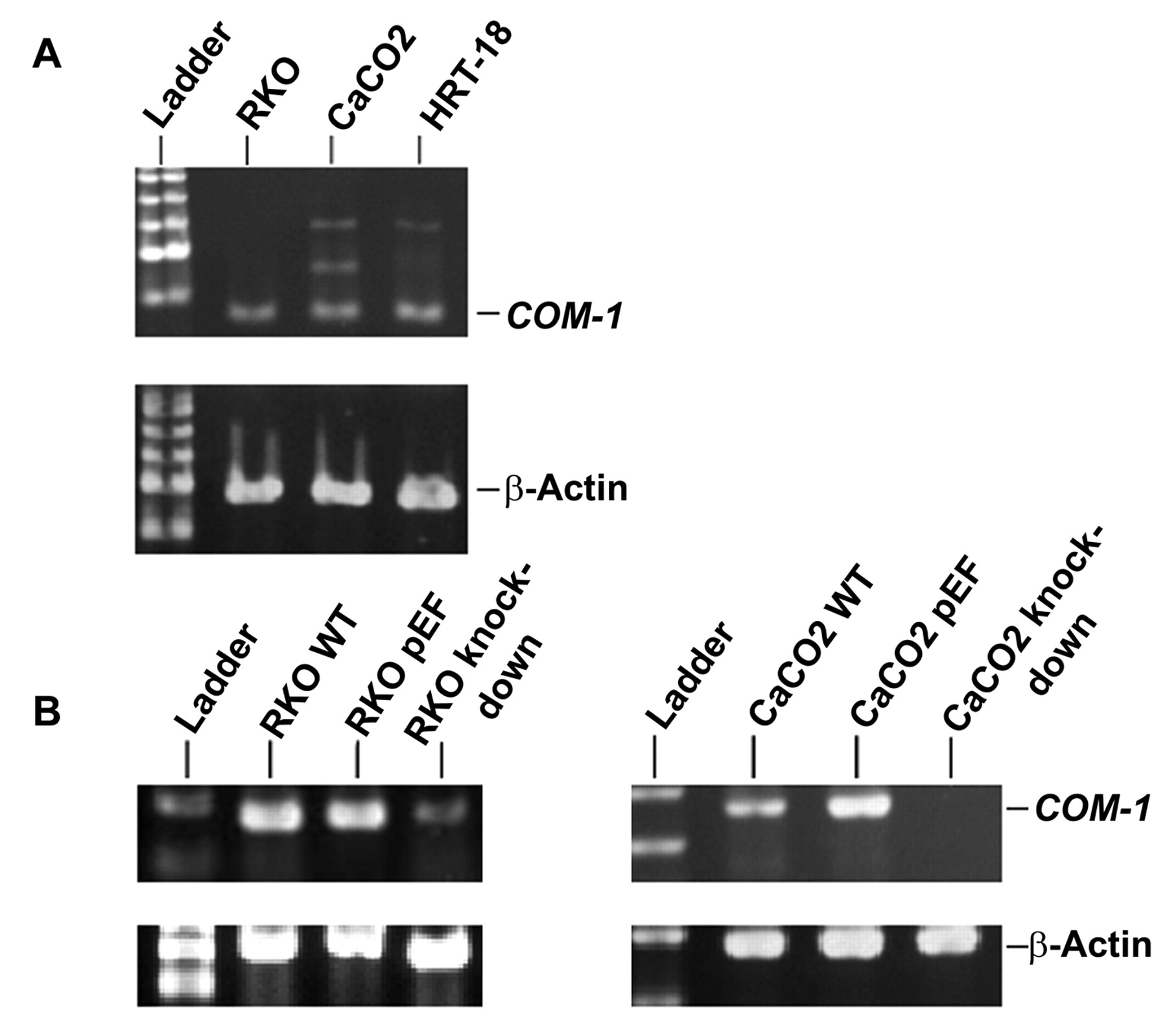
The manipulation of candidate of metastasis-1(COM-1) gene expression in colorectal cancer cells. A: Expression of COM-1 in RKO, CaCO2 and HRT-18 cells. B: Knock-down of COM-1 in RKO and CaCO2 cells.
Evaluation of apoptosis. RKO and CaCO2 cells were seeded at 1×105 cell and 1.5×105 cells in 25 cm3 culture flasks. The medium used consisted of 3.0 μM ciglitazone. A negative control was prepared by incubating cells in the absence of ciglitazone. After three days, the cells were harvested and washed twice in cold borate buffered saline (BBS). The cells were resuspended in 100 μl 1x annexin V-binding buffer (Sigma-Aldrich Ltd). 1 μl of the 100 μg/ml working solution and 5 μl of the fluorescein isothiocyanate (FITC) annexin V were added to each 100 μl of cell suspension. The cells were incubated at room temperature for 15 min. After that, 400 μl of 1× annexin-V binding buffer were added, mixed gently and the cells kept on ice. As soon as possible, the stained cells were analyzed by flow cytometry to asses the percentage of apoptotic cells.
Statistical analysis. Statistical analysis was carried out using student t-tests for all data following a normal distribution (Excell, Microsoft, USA) and expressed as mean plus/minus standard error.
Results
Knock-down of COM-1 in human colorectal cells lines. We confirmed the expression of COM-1 in three human colorectal cell lines, RKA, CaCO2 and HRT-18 using RT-PCR (Figure 1A). After construction of a ribozyme transgene, COM-1 expression was knocked-down and this was confirmed in RKA and CaCO2 cells using RT-PCR (Figure 1B). β-Actin was used as a control for cDNA consistency.
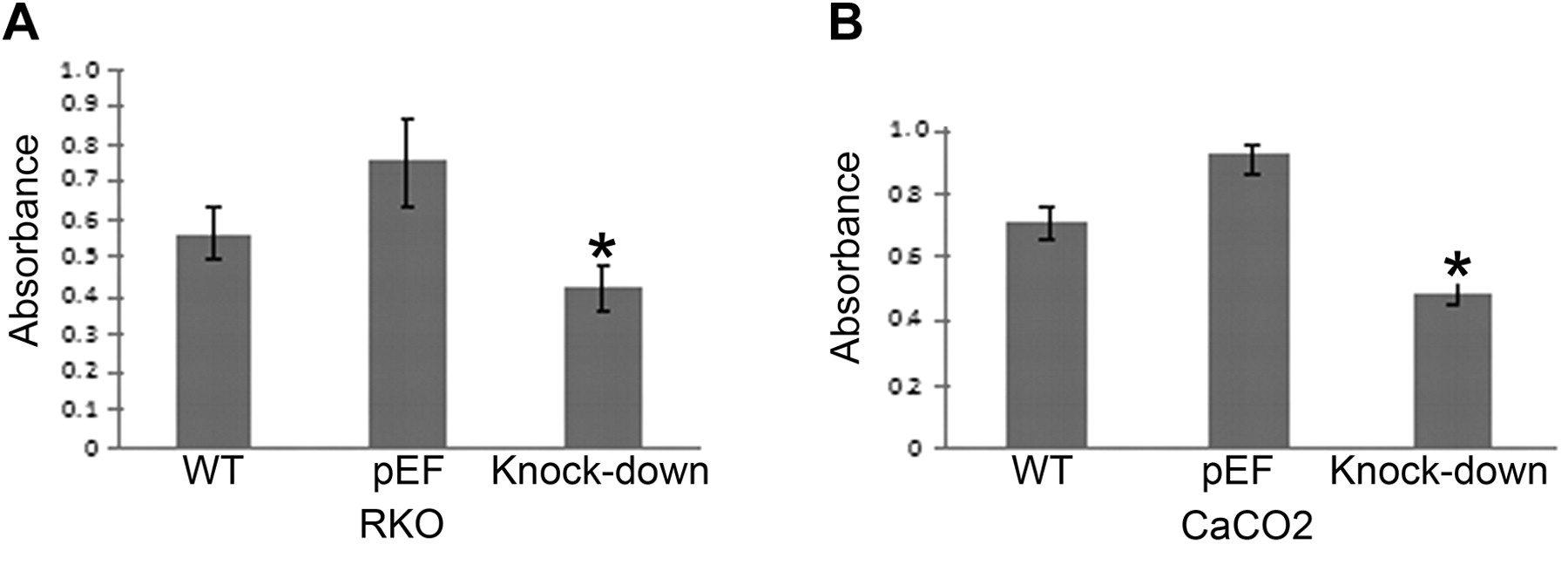
The effect of COM-1 knock-down on cell growth. Growth assays were carried out to assess the effect of COM-1 knock down on colorectal cancer cell growth. Both RKO and CaCO2 cell lines exhibited significantly reduced growth over 3 days after COM-1 knock-down (p<0.01), as shown in Figure 2A and B (RKO and CaCO2 cell lines, respectively).
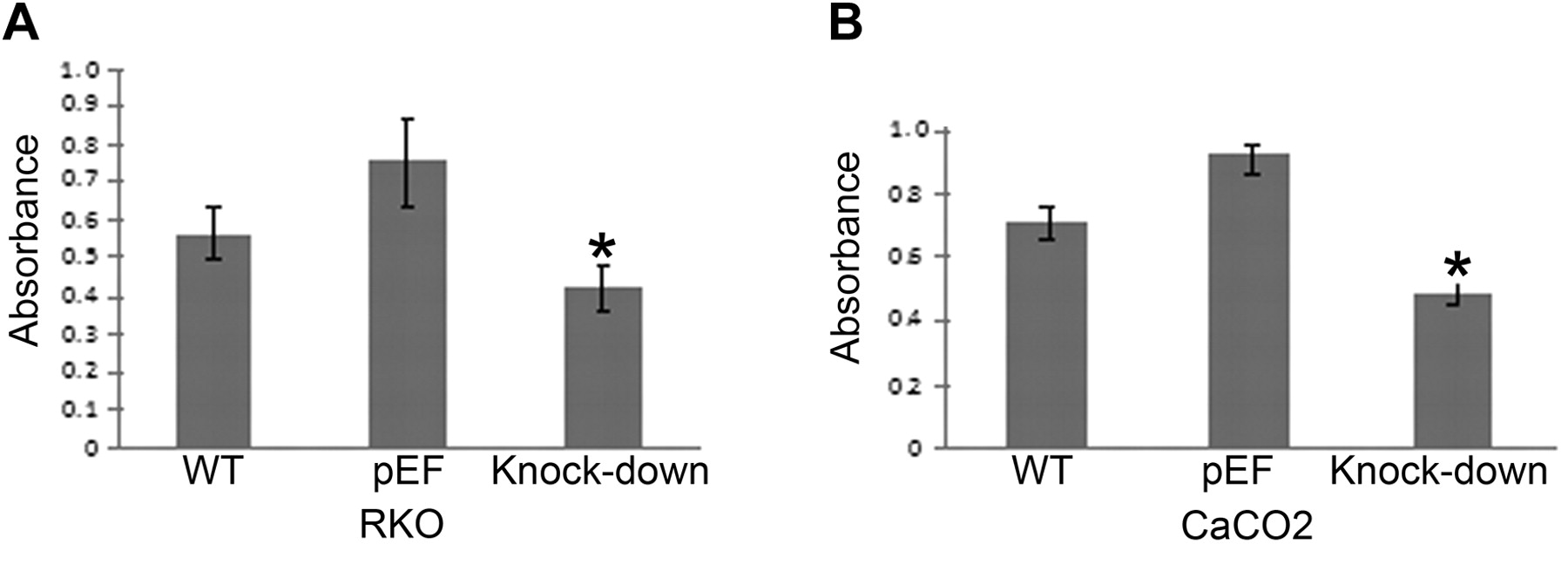
Growth of COM-1 knock-down cells. Both RKO (A) and CaCO2 (B) cell lines exhibited significantly reduced growth over 3 days after COM-1 knock-down (p<0.01 vs. pEF cells).

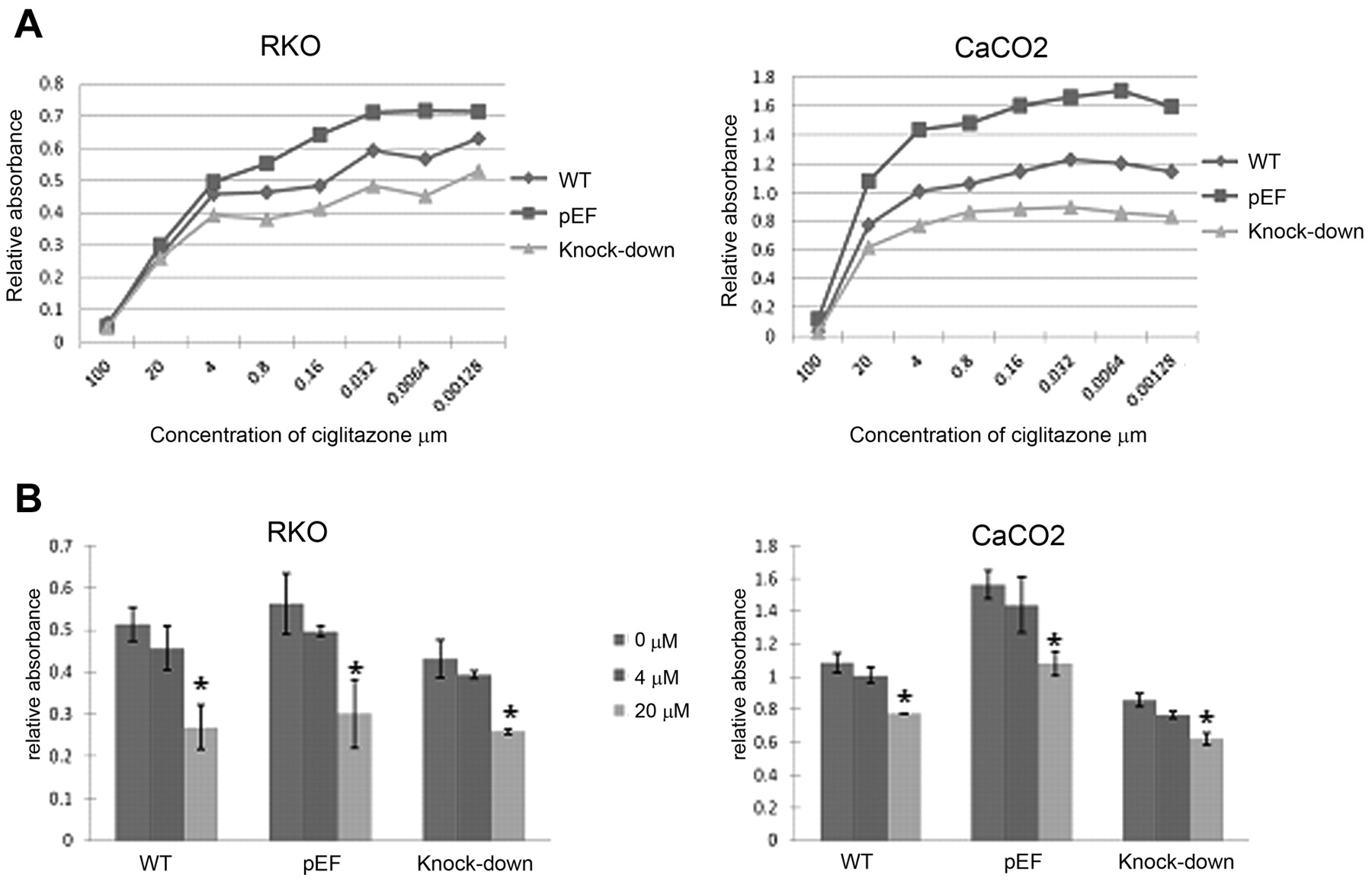
Assessment of growth rates of colorectal cancer cells. A: Growth of COM-1 knock-down cells cultured with ciglitazone in different concentrations from 100 μM to 0.00128 μM. B: 20 μM ciglitazone inhibits RKO and CaCO2 cell growth (compared to controls, p<0.05). Cigltiazone at 4 μM inhibits cell growth to some extent, but not statistically significantly.
The effect of PPARγ agonists on growth of COM-1 knock down cells. As COM-1 is believed to be a tumour suppressor in human prostate cancer cells and that the potential tumour suppressive effect of COM-1 is at least partly via its interaction with PGC-1, the PPARγ coactivator. we used the PPARγ agonists ciglitazone and BADGE to identify the relationship between COM-1 and PPARγ. The colorectal cancer cells were treated with ciglitazone or BADGE for three days at different concentrations ranging from 100 μM to 0.00128 μM. The 50% effective concentration (EC50) of ciglitazone was 3.0 μM and the 50% inhibitory concentration (IC50) was 5.5 μM in the RKO and CaCO2 wild-type cells. We used absorbance to determine the number of the cells that had grown in 96-well plates. From Figure 3A, it can be seen that ciglitazone reduced the growth of all cells over three days. The greatest effect was observed at 100 μM (p<0.05) (Figure 3B). The combination of ciglitazone and COM-1 knock-down exhibited an additive effect in reducing growth. The COM-1 knock-down cells also grew slower than both the control cells and wild-type even after treatment with ciglitazone. We then sought to determine if a similar effect would also occur using BADGE. Again, similar concentrations were used. This time however, there was no effect on growth with the agonist BADGE (Figure 4A and B) not even in control cells.

Effect of bisphenol A diglycidyl ether (BADGE) on cell growth. A: Growth of COM-1 knock-down cells cultured with BADGE at different concentrations from 100 μM to 0.00128 μM. B: BADGE had little effect on RKO and CaCO2 cell growth at 4 or 20 μM.
The effect of COM-1 knock-down on cell migration. As COM-1 is thought to be a regulator of metastasis, we continued our investigation into its role in human colorectal cells by looking at a key event in the metastatic cascade, cell migration. As shown in Figure 5, knock-down of COM-1 had little effect on the migratory behaviour of either RKO and CaCO2 cells as assessed using ECIS technology (Figure 5A) and wounding assays (Figure 5B).
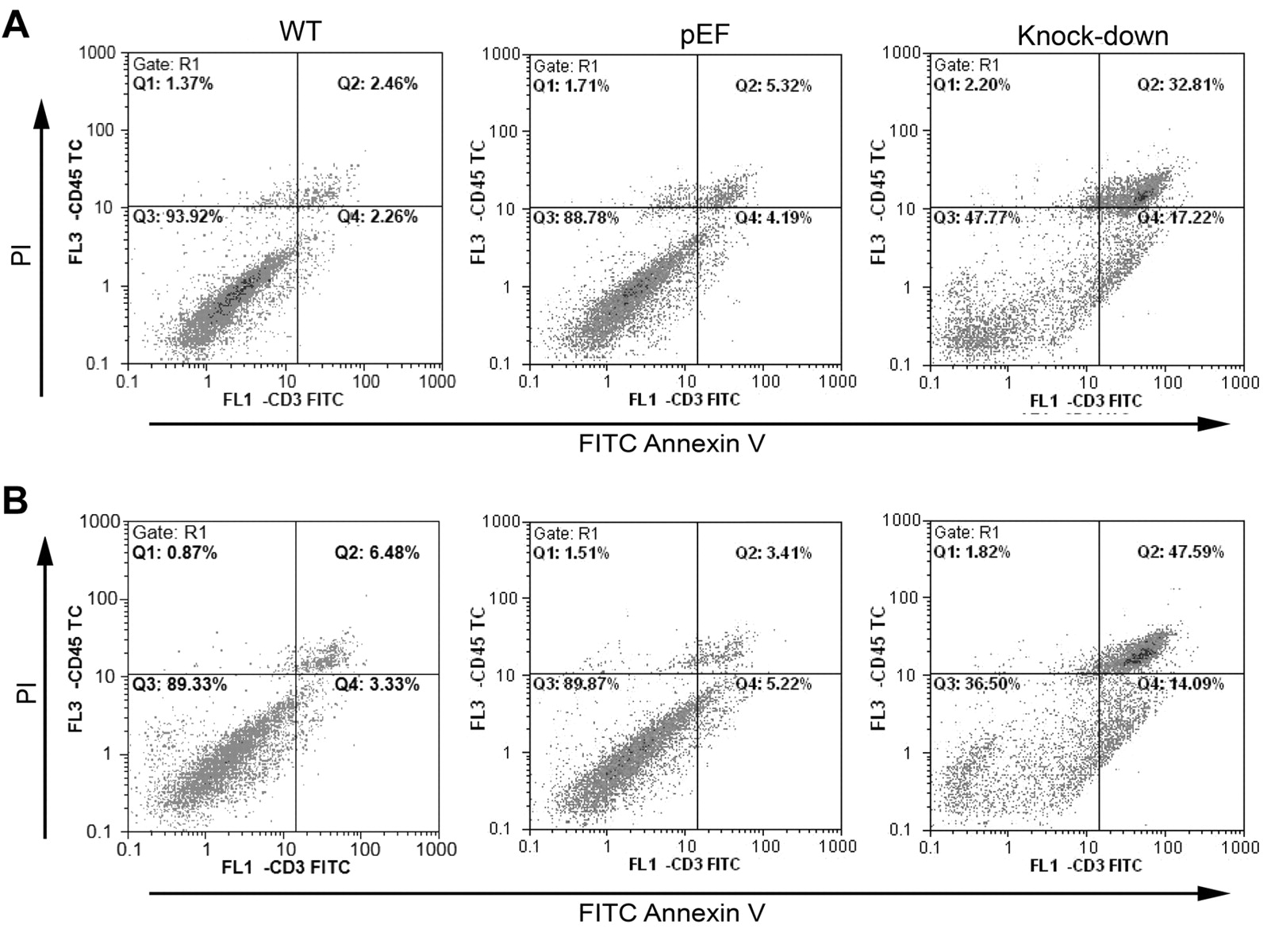
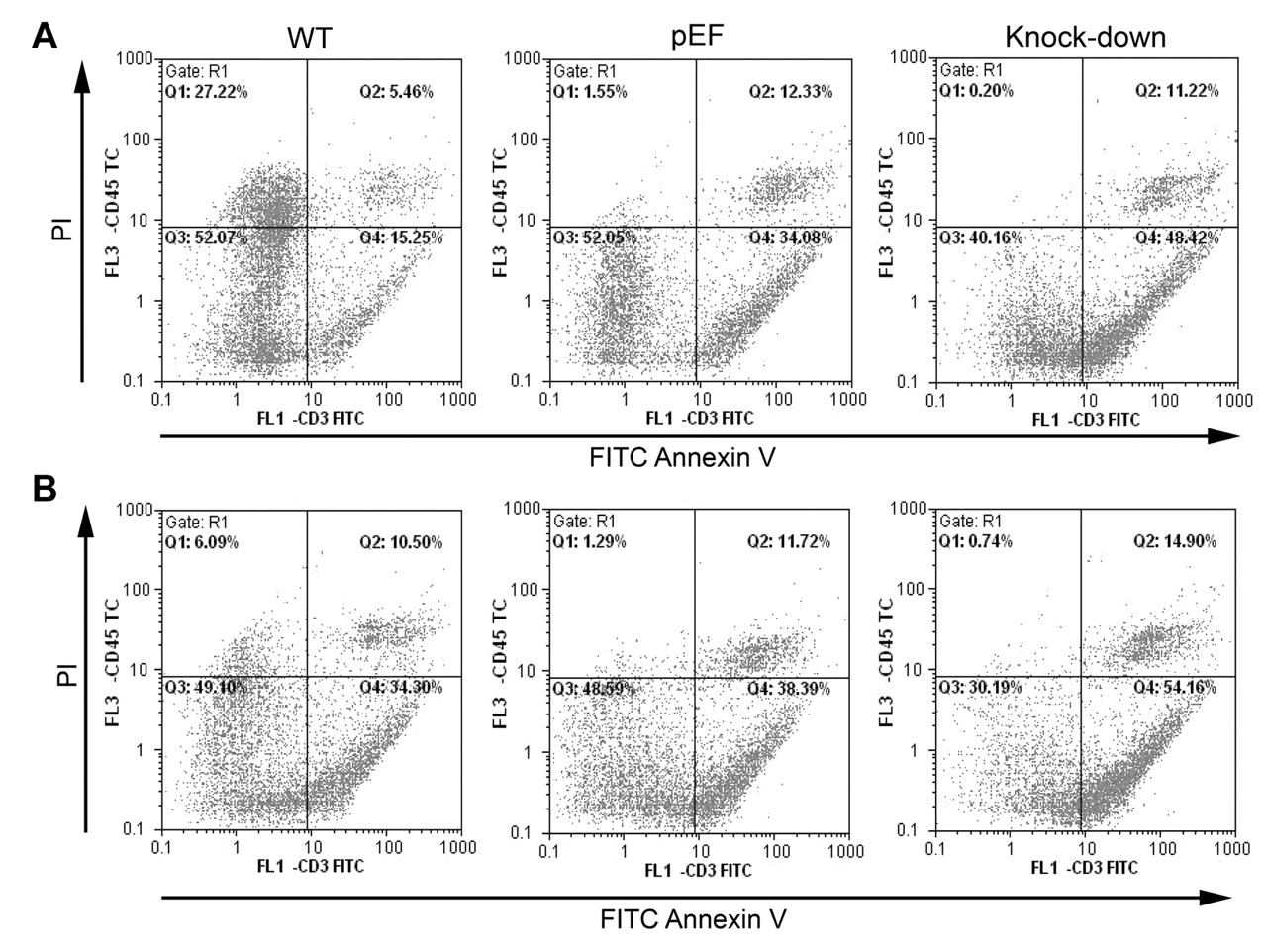
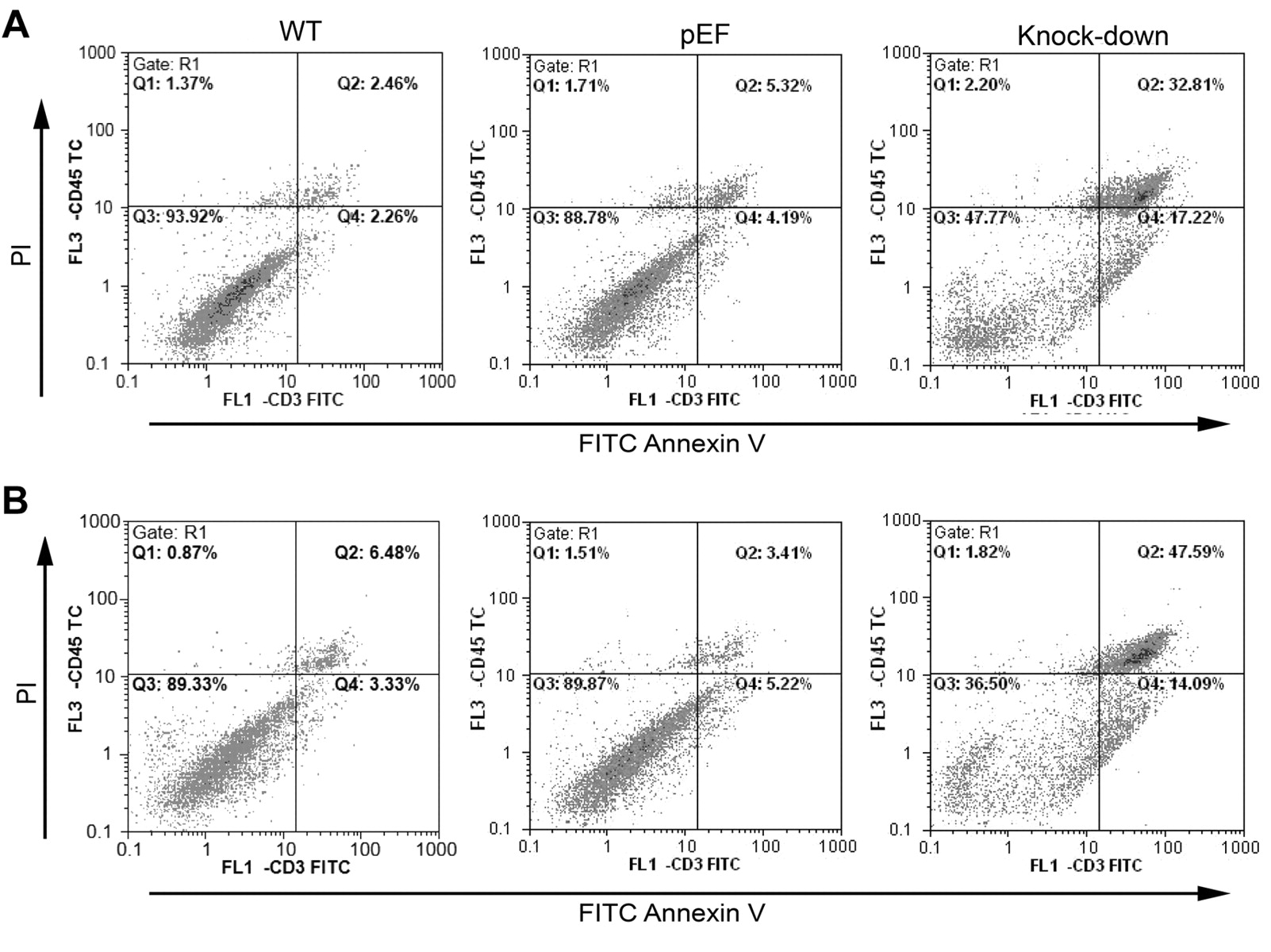
The effect of COM-1 knock down on apoptosis. To determine if the reduction in cell growth was a consequence of cell death and apoptosis, we carried out apoptosis assays. Figure 6Ashows that there was an increase in apoptosis and RKO COM-1 knock-down cell death (48.42% cells apoptotic) when compared to control (34.08%) and wild-type cells (15.25%). This apoptotic effect was increased after treatment with ciglitazone (Figure 6B). This was also the case for the COM-1 knock down CaCO2 cells, where 17.22% of the cells were apoptotic as compared to 4.19% of the pEF control cells and 2.26% of the wild-type cells (Figure 7A). Again, ciglitazone treatment accentuated the effect of COM-1 knock-down (Figure 7B). From this, we can see that apoptosis must be contributing to the reduction in cell growth brought about by COM-1 knock-down. Ciglitazone also induces apoptosis in wild-type and control cells and increases apoptosis in COM-1 knock-down cells.

Cell migration of COM-1 knock-down cells. Both RKO and CaCO2 cells exhibited no difference in motility (p>0.05) when COM-1 was knocked down as assessed using both ECIS (A) and wounding assays (B).
Discussion
In this study we have revealed the in vitro effect of COM-1 in colorectal cancer. When COM-1 was knocked out, colorectal cancer cells grew more slowly than did control cells. COM-1 is thus able to effect changes in cell growth; however from our work and from literature, it appears that this effect is dependent on the cell type used, with COM-1 being able to inhibit or promote the growth of cells. COM-1has been shown to inhibit the growth of human pancreatic cancer cells (7) and may also have a suppressive role in breast cancer (8) and prostate cancer (9). In contrast, some studies have described the reverse. Gustavo et al. have showed that COM-1 overexpression promoted cell growth in the renal cell line COS-7 and in the pancreatic acinar cell line, AR4-2J (10). In human pancreatic cancer tissues, overexpression of COM-1 is inversely related to apoptosis. Additionally, in other neoplasms such as thyroid cancer, COM-1 overexpression may be related to progression and metastasis (5). In colorectal cancer, we have previously shown that that COM-1 may be a tumour suppressor in human colorectal cancer after studying its expression in cancer and normal tissues (6). One of the reasons for this disparity in function may be due to the specificity of is action in cells and/or tissues. Different cancer cell lines and indeed different experimental methods may lead to different cell growth results. It is likely however, that the difference is due to the complexity of COM-1 functions. COM-1 is thought to act as a transcription regulator and accordingly can regulate downstream transcriptional activities, through which it can increase or suppress cell growth rate. Moreover, some upstream transcriptional factors can also regulate the expression of COM-1 itself. CCAAT/enhancer binding proteinα (C/EBPα) and C/EBPβ were described as being able to regulate COM-1 transcription (11). Hence, the diverse function of COM-1 could be due to a number of transcriptional factors. Furthermore, COM-1 is also a stress protein, both in vivo and in vitro, where a number of stress activators have been shown to activate COM-1 transcription. For instance, inhibitory signals (12), anticancer drugs (13) and starvation (14) can activate the expression of COM-1. The function of COM-1 is thus complex and in order to explain these functions and the role of COM-1 in cancer development better, in vivo assays are essential.
PPAR-γ is a nuclear hormone receptor and plays an important role in energy metabolism, adipocyte differentiation, fat storage and inflammation. Increasing evidence has shown that PPARγ has an anti-neoplastic effect in many types of tumours, including breast cancer (15), lung cancer (16), prostate cancer (17), human multiple myeloma (18) and colorectal cancer (19). PPARγ is activated by binding to its ligands. Importantly, treatment with the PPARγ agonist ciglitazone, augments its effects. 15-Deoxy-Δ 12,14-prostaglandin J2 (15d-PGJ2) is the most potent endogenous ligand for PPARγ (20). Currently, thiazolidinediones (TZDs) are widely used as synthetic ligands for PPARγ. There are ciglitazone, rosiglitazone, troglitazone, pioglitazone and BADGE. In our study, we used ciglitazone and BADGE as PPARγ agonists to identify the potential relationship between COM-1 and PPARγ. In our study, colorectal cancer cells were respectively treated with ciglitazone and BADGE over 3 days with different concentrations. We observed that ciglitazone did suppress cell growth at some concentrations. In addition, COM-1 knock-down cells also grew slower than did control transfected cells and wild-type cells when treated with ciglitazone, from which we conclude that PPARγ had no effect on COM-1. Moreover, previous work (8) conducted by our laboratory found that PGC-1, but not PPARγ was one of the few proteins that co-precipitated with COM-1. These results were consistent in both colorectal cancer cell lines. However, BADGE had little or no effect, unlike ciglitazone.

Effect of ciglitazone on cell growth. RKO cells were cultured in normal medium (A) and medium with 3 μM ciglitazone (B) for 3 days. A: There were more apoptotic (48.42%) and dead (11.22%) COM-1 knock-down cells than wild-type (WT) and pEF control cells. B: When treated with ciglitazone for 3 days, more cells were in the state of apoptosis and death than those not treated with ciglitazone respectively. COM-1 knock down still increased apoptosis.
COM-1 protein is highly overexpressed in some type of cancer at the late stage or when metastasis occurs (1, 4-5). Hence is it possible that COM-1 plays a positive role in cancer progression. Since cell migration is a highly integrated, multi-step process that plays an important role in the progression of cancers, we investigated the effect of COM-1 knock-down in order to observe any changes in cell motility. COM-1 knock-down cells exhibited no difference in cell motility. This could mean that during cancer progression, COM-1 has no effect on motility. To our knowledge, this is the first study to examine the effect of COM-1 on motility in vitro, especially in colorectal cancer cells. However, the question as to whether COM-1 is pro- or anti -metastatic is still unanswered. Ree et al. considered COM-1 to be a metastasis promoting gene by establishing tumour metastases in mouse models (1). Since then, Ito et al. analyzed thyroid cancer clinical data and concluded that COM-1 expression and tumour lymphatic metastasis were significantly related, which supported the idea that the molecule is metastasis-related (5). On the other hand, it has been suggested that COM-1 plays a role in early-stage breast cancer, but has no role in late stage and metastasis (1). From these in vivo assay and clinical data, it is still difficult to draw a final conclusion as to whether COM-1 and metastasis are related or not. Nevertheless, we can confirm that from our migration assay COM-1 has no effect on colorectal cancer cell motility in vitro.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Effect of ciglitazone on apoptosis. CaCO2 cells were cultured in normal medium (A) and medium with 3 μM ciglitazone (B) for 3 days. A: There were more apoptotic (17.22%) and dead (32.81%) COM-1 knock-down cells than wild-type (WT) and pEF control cells. B: When treated with ciglitazone for 3 days, more cells were in the state of apoptosis and death than those not treated with ciglitazone respectively. COM-1 knock-down again increased apoptosis.
As COM-1 does have an effect on cell growth, the question arises as to how. Apoptosis is programmed cell death, which can regulate cell growth, development and differentiation (21). In our study, COM-1 knock-down cells were more apoptotic than control and wild-type cells. Su et al. considered that overexpression of COM-1 is inversely correlated with apoptosis in pancreatic cancer (4). Kong et al. used an RNAi method to prove that COM-1 silencing increased autophagy and apoptosis in cardiac cells (22). Giroux et al. suggested that in pancreatic cancer cells, a large part of gemcitabine-induced apoptosis resulted from the inhibition of the constitutive antiapoptotic activity of COM-1 (23). Malicet et al. proved that COM-1 bound the antiapoptotic protein prothymosin α, forming a complex which then resulted in important changes in the secondary and tertiary structures of the proteins (24). They discovered that COM-1 participates in the process of anti apoptosis together with prothymsin α. In our study, when treated with PPARγ over 3 days, we found that more cells were dead and apoptotic than control cells, and that the COM-1 knockdown cells were the most apoptotic cells. The apoptosis results were in agreement with those of cell growth. Thus, when COM-1 was knocked down, more cells were apoptotic which led to inhibition of cell growth. Ciglitazone also induced the process of apoptosis in both control cells and COM-1 knock down cells.
From our results, we can conclude that in RKO and CaCO2 colorectal cancer cell lines, COM-1 is an anti-apoptotic gene and a cell growth promoter. PPARγ agonist further inhibited COM-1 knock-down–induced cell growth and promoted its apoptosis. In the metastatic cascade, COM-1 appears to play no role in motility/migration of colorectal cancercells.
Acknowledgements
Dr Xin Li is a recipient of the China Medical Scholarship of Cardiff University. We wish to thank the Albert Hung Foundation for supporting this work. We would also like to thank Cancer Research Wales for sponsoring Dr Tracey A. Martin.
- Received February 3, 2012.
- Revision received March 15, 2012.
- Accepted March 16, 2012.
- Copyright© 2012 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved