

Abstract
Background/Aim: The phenomenon of membrane vesicle-release by neoplastic cells is a growing field of interest in cancer research, due to their potential role in carrying a large array of tumor antigens when secreted into the extracellular medium. In particular, experimental evidence show that at least some of the tumor markers detected in the blood circulation of mammary carcinoma patients are carried by membrane-bound vesicles. Thus, biomarker research in breast cancer can gain great benefits from vesicle characterization. Materials and Methods: Conditioned medium was collected from serum starved MDA-MB-231 sub-confluent cell cultures and exosome-like vesicles (ELVs) were isolated by ultracentrifugation. Ultrastructural analysis of ELVs was performed by transmission electron microscopy (TEM) and the purity of fraction was confirmed by western blotting assays. Proteomic profile of ELVs was carried out by 2 D-PAGE and protein identification performed by MALDI-ToF Mass Spectrometry. Results: On the basis of ultrastructural and immunological characterization, the isolated vesicles have been classified as exosome-like vesicles (ELVs). The proteomic investigation showed a distinctive protein profile of the ELVs, in comparison to the whole cell lisates (WCL) proteome, which could be instrumental for cancer progression. The proteins were clustered into functional categories, according to the current bioinformatics resources and a Venn diagram was constructed based on these clusters. Conclusion: It is reasonable to assume that vesicle production allows neoplastic cells to exert different effects, according to the possible acceptor targets. For instance, vesicles could potentiate the malignant properties of adjacent neoplastic cells or activate non-tumoral cells. Moreover, vesicles could convey signals to immune cells and surrounding stroma cells. The present study may significantly contribute to the knowledge of the vesiculation phenomenon, which is a critical device for trans cellular communication in cancer.
- Breast cancer
- extracellular vesicles
- protein biomarker
- 2D-PAGE
- proteomic profiling
- MALDI-ToF
- mass spectrometry
- MDA-MB-231 cells
The phenomenon of membrane release in the extracellular medium has long been known and was firstly described by Paul H. Black in normal and cancer cells (1). More recently, it has been observed that the extracellular environment may contain a large number and a variety of membrane-coated vesicles, hence the term ‘extracellular vesicles’ (EVs) was suggested to include all the above (2).
To distinguish the different type of vesicles, classification criteria based on their different origin, biogenesis and function have been proposed. Currently, major vesicle populations are grouped as exosomes, microvesicles and apoptotic bodies. The expression ‘exosome’ refers to vesicles of 50-100 nm in diameter, generated by an exocytotic process. Indeed, the exosomes represent a rather homogeneous family of small vesicles which are generated from inward budding of the limiting membrane of late endosomal compartments. The latter forms intra-luminal vesicles inside structures known as multivesicular bodies (MVBs). MVBs eventually fuse with the cell membrane, releasing their content – namely exosomes – into the extracellular space (3, 4). Vesicles having similar morphology and size and unknown biogenesis are defined as exosome-like vesicles (ELVs).
The microvesicles (MVs) are larger vesicles (0.1-2 μm diameter) released by the outward budding of the cell membrane. MVs were first described by Chargaff and West in 1946 as a precipitable factor in platelet-free plasma with thromboplastic potential (5). Over the years they have been principally characterized as products of endothelial cells, platelets and other blood and tumor cells. Key functions of MVs include pro-coagulant activity (6), involvement in the pathogenesis of rheumatoid arthritis (7), tumor invasion (8, 9), oncogenic transformation (10), induction of angiogenesis (11-13) and feto-maternal communication (14).
The category of apoptotic bodies includes the largest vesicle population (1-5 μm in diameter) (approximately the size range of platelets) (2). The term ‘apoptotic body’ was introduced by Kerr in 1972 (15) because they are mostly released as blebs from cells undergoing apoptosis. These kind of vesicles are characterized by phosphatidylserine externalization and may contain fragmented DNA (16).
The growing interest in exosomes, in cancer research, arises from their potential role in carrying a large array of tumor antigens when secreted by neoplastic cells. For example, melanoma-derived exosomes contain the highly immunogenic antigens MelanA/Mart-1 and GP100 (17). Similarly exosomes shed by colon carcinoma cells express carcino-embryogenic antigen (CEA) and human epidermal growth factor receptor-2 (HER2), which can also be detected in vesicles secreted by breast carcinoma (18, 19). Likewise, membrane vesicles from two breast carcinoma cell lines, MCF-7 and 8701-BC, have been shown to carry most antigens expressed on the cell surface (20) such as: β1, α3 and α5 integrin chains, tumor-associated antigens, HLA class I molecules, matrix metalloproteinase-9 (MMP-9), and tissue inhibitor of metalloproteinase-1(TIMP-1).
This experimental evidence shows that at least some of the tumor markers detected in the blood circulation of patients with mammary carcinoma are carried by membrane vesicles. Thus, biomarker research in breast cancer can gain great benefits from vesicle characterization. Recently, proteomic analysis has become the technique of choise for characterizing EVs, because a wider knowledge on EV protein content can help in understanding their potential roles in vivo, and also because it could allow identification of new tumoral markers with diagnostic value.
In this study, we report the isolation of ELVs from serum-free culture medium of a highly invasive breast cancer cell line, MDA-MB-231, chosen as a model system for vesicle release. This cell line, derived from the malignant pleural effusion of a patient with invasive breast cancer, has been widely studied and characterized, but the nature and the composition of shed vesicles has not been investigated. Moreover, this cell line has been shown to shed vesicles even in absence of serum (21), thus allowing the collection of uncontaminated vesicles. Indeed it is known that the phenomenon of vesicle shedding in vitro by several cell types is also influenced by culture conditions, and especially by the presence of serum (22).
The purity of our ELV fraction was confirmed by electron microscopy and western blotting assays, before investigation their proteomic profile, by 2-D-PAGE and mass spectrometry. The results showed a distinctive protein profile of the vesicles in comparison with the whole cell proteome, which could be instrumental for cancer progression.
Materials and Methods
Cell culture. Breast cancer MDA-MB-231 cells, originally derived from a pleural effusion, were obtained from the American Type Culture Collection (ATCC, Sesto San Giovanni, Milan, Italy). Cells were seeded at a density of 104 cells/cm2 and grown in RPMI-1640 culture medium (CELBIO, Pero, Milan, Italy), with L-glutamine (2 mM) and antibiotics (100 units/ml penicillin and 100 units/ml streptomycin), supplemented with 10% fetal bovine serum (CELBIO) and grown in a humidified incubator with 5% CO2 in air at 37°C.
Sample preparation. Cells were grown in the presence of serum until 80-90% confluence. Then they were washed twice in phosphate-buffered saline (PBS) and fresh medium depleted of serum was added. After 4 h starvation, the conditioned medium was discarded and fresh medium was added without serum; conditioned medium was then collected 24 h later. After washing with cold PBS, cells were carefully scraped and incubated on ice for 30 min with RIPA buffer (50 mM Tris, pH 7.5, 0,1% NP40, 0,1% deoxycholate, 150 mM NaCl and 4 mM EDTA) and a mixture of protease inhibitors [0.01% aprotinin, 10 mM sodium pyrophosphate, 2 mM sodium orthovanadate and 1 mM phenylmethylsulfonyl fluoride (PMSF)]. The whole cellular lysate was centrifuged at 14,000 rpm (16,000 ×g) for 8 min in order to clear cell debris and were then stored at −80°C. Protein concentration in the cellular extracts was determined using the Bradford method (23).
Vesicle collection. Conditioned media from replicate cultures were centrifuged at 2,000 ×g for 10 min, 4,000 ×g for 15 min and 15,000 ×g (JA 20 Rotor, Beckman, Cassina De’ Pecchi, Milan, Italy) for 30 min at 4°C to remove cellular debris. Each supernatant was then ultracentrifuged at 100,000 ×g (Ti 60 Rotor, Beckman) for 90 min at 4°C (24) and the pellet was collected. The vesicle fractions were re-suspended in PBS and stored at -80°C until use. Protein concentration was determined using the Bradford method.
Transmission electron microscopic (TEM) analysis. Five microliter aliquots of re-suspended vesicle fraction were placed onto support grids (Cu 200M), and allowed to adsorb for 60 s. Grids were washed twice in double-distilled water, and vesicles were then stained for 10 s in 2% aqueous solution of uranyl acetate. The size and morphology of the particles were examined using an FEI Morgagni transmission electron microscope.
The number of vesicles in each field was extrapolated after analyzing three images. Each field had the following sizes: 7.2 μm × 5.7 μm. The vesicles in each field were respectively 139, 132 and 131, with a mean value of 134 vesicles for each field.
Western blotting. For immune detection the polyacrylamide 1D gels (SDS-PAGE) of putative ELVs fraction and whole-cell lysates, were electrotransferred onto nitrocellulose membrane (HyBond ECL; Amersham, Uppsala, Sweden) and stained with Ponceau S (Sigma Aldrich, St. Louis, Missouri, United States). The membranes were then probed with one of the following monoclonal antibodies (mAb): anti-beta1 integrin C27 rat mAb (1:5000, Wen-Tien Chen Lab., Stony Brook, New York, United States); anti Heat shock cognate 70 (HSC70) mouse mAb (1:2500; Santa Cruz, Heidelberg, Germany); anti-cytochrome c clone 7H8.2C12 mouse mAb (1:500; BD Pharmingen, New Jersey, United States); anti Lysosome-associated membrane glycoprotein 1 (LAMP1) mouse mAb (1:50; Santa Cruz). Following incubation with the appropriate peroxidase-linked antibody [(horseradish peroxidase-conjugated anti-rat IgG (1:20000; Sigma Aldrich); horseradish peroxidase-conjugated anti-mouse IgG (1:5000; Sigma Aldrich)], the reaction was revealed by the ECL detection system, using high performance films (Hyperfilm ECL; Amersham).
Two dimensional gel electrophoresis. Both the vesicle fractions and the cell lysates were submitted to extensive dialysis against ultrapure distilled water at 4°C and lyophilized. Dried samples were solubilized in a buffer containing 4% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS), 40 mM Tris, 65 mM 1, 4 dithioerythrol (DTE) and a trace of bromophenol blue in 8 M urea. Aliquots of 45 μg (analytical gels) or 1.5 mg (preparative gels) of total proteins were separately mixed with 350 μl of rehydration solution containing 8 M urea, 2% CHAPS, 10 mM DTE and 0.5% carrier ampholytes (Resolyte 3.5-10; Amersham), and applied for isoelectric focusing (IEF) using commercial sigmoidal IPG strips, 18 cm long with pH range 3.0-10; (Bio-rad, Segrate-Milan, Italy). The IEF was carried out by linearly increasing the voltage from 200 to 3500 V during the first 3 h, after which focusing was continued at 8000 V for 8 h. After the run, the IPG strips were equilibrated with a solution containing 6 M urea, 30% glycerol, 2% Sodium Dodecyl Sulphate (SDS), 0.05 M Tris-HCl, pH 6.8 and 2% DTE for 12 min, in order to re-solubilize the proteins and reduce the disulphuric bonds. The –SH groups were then blocked by substituting the DTE with 2.5% iodoacetamide in the equilibrating buffer. The focused proteins were then separated on 9-16% linear gradient polyacrylamide gels (SDS-PAGE) with a constant current of 20 mA/gel at 10°C and the separated proteins were visualized by ammoniacal silver staining.
Image acquisition and data analysis. Silver-stained gel images were digitized using computing densitometry and analyzed with ImageMaster 2D PLATINUM software (Amersham Biosciences, Sweden). Gel calibration was carried out using an internal standard and the support of the ExPaSy molecular biology server, as described elsewhere (25). Quantitative variations in the protein expression levels were calculated as the volume of the spots (i.e. integration of optical density over the spot area). In order to correct for differences in gel staining, the spot volumes relative to the sum of the volume of all spots on each gel (%Vol) were calculated by the software.
Protein identification by N-Terminal microsequencing. N-Terminal microsequencing was performed by automated Edman degradation in a protein sequencer (Procise 419; Applied Biosystems), as previously described (26).
In-gel digestion and MALDI-ToF analysis. Mass spectrometric sequencing was performed by Voyager DE-PRO (Applied Biosystems) mass spectrometer as described elsewhere (27). Briefly, proteins were digested using sequencing-grade trypsin (20 μg/vial). The tryptic peptide extracts were dried and re-dissolved in 10 μl of 0.1% trifluoroacetic acid (TFA). The matrix, R-cyano-4-hydroxycinnamic acid (HCCA), was purchased from Sigma-Aldrich. A saturated solution of HCCA (1 μl) at 2 mg/200 μl in CH3CN/H2O (50:50 v/v) containing 0.1% TFA was mixed with 1 μl of peptide solution on the MALDI plate and left to dry. MALDI-ToF mass spectra were recorded in the 500-5000 Da mass range, using a minimum of 100 shots of laser per spectrum. Delayed extraction source and reflector equipment allowed sufficient resolution to consider MH ± of monoisotopic peptide masses. Internal calibration was carried out using trypsin autolysis fragments at m/z 842.5100, 1045.5642, and 2211.1046 Da. Peptide mass fingerprinting was compared to the theoretical masses from the Swiss-Prot or NCBI sequence databases using Mascot (http://www.matrixscience.com/). Typical search parameters were as follows: 50 ppm of mass tolerance, carbamidomethylation of cysteine residues, one missed enzymatic cleavage for trypsin, a minimum of four peptide mass hits was required for a match, methionine residues could be considered in oxidized form.
In gel digestion and LC-MS/MS analysis. The gel slices were washed with 50% acetonitrile/0.1 M ammonium bicarbonate for 4 h and dried. The proteins were reduced prior to digestion with 10 mM Dithiothreitol (DTT) for 2 h at 37°C and alkylated with iodoacetamide (50 mM final concentration) for 15 min in the dark at room temperature. The gel spots were washed again with 50% acetonitrile/0.1 M ammonium bicarbonate for 2 h. Digestion was carried out with 125 ng trypsin (Sequencing Grade, Promega, Madison, Wisconsin, USA) in 0.05 M ammonium bicarbonate overnight at 37°C. The peptides in the supernatant were collected and the gel pieces were extracted with 0.1% acetic acid/ 50% acetonitrile and the extract was pooled with the tryptic peptides. The digest was dried in a Speed Vac and re-dissolved in 50 μl 0.1% TFA. The tryptic digests were analyzed by a capillary liquid chromatography tandem MS (LC-MS/MS) using a set up of a ProteoCol trap C-18 column (0.15 × 10 mm, 3 μm particle size, 300Å; SGE Analytical Science, Victoria, Australia) and a separating column (0.1 mm × 10 cm) that had been packed with Magic 300Å C18 reverse-phase material (5 μm particle size; Swiss Bioanalytics, Birsfelden, Switzerland). The columns were connected on line to an Orbitrap FT hybrid instrument (Thermo Finnigan, San José, CA, USA). The solvents used for peptide separation were 0.1% acetic acid in water (solvent A) and 0.1% acetic acid and 80% acetonitrile in water (solvent B). Peptides were injected via a 2 μl loop onto the trap column with the capillary pump of an Agilent 1200 system (Agilent, Cernusco, Milano, Italy) set to 5 μl/min. After 15 min, the trap column was switched into the flow path of the separating column. A linear gradient from 2% to 35% solvent B in solvent A in 60 mins was delivered with an Agilent 1200 nano pump at a flow rate of 300 nl/min. After 60 min the percentage of solvent B was increased to 60% in 10 min and further increased to 80% within 2 min. The eluting peptides were ionized at 1.7 kV. The mass spectrometer was operated in a data-dependent fashion. The precursor scan was carried out in the Orbitrap set to 60,000 resolution, while the fragment ions were mass analyzed in the Linear trap quadrupole (LTQ) instrument. A top five method was run so that the five most intense precursors were selected for fragmentation. The MS/MS spectra were then searched against the NCBI non-redundant database, version from February 26th 2010, using TurboSequest software (28). The databank was searched with Bioworks version 3.3.1. SP1 by setting the precursor ion tolerance to 10 ppm, while the fragment ion tolerance was set to 0.5 Da. Cleavage rules were set to fully enzymatic – cleaves at both ends, allowing two missed cleavages. Post filtering was set to the following parameters: ΔCN, 0.1; Xcorr versus charge state was 1.50 (1+), 2.00 (2+), 2.50 (3+); peptide probability, 0.01; protein probability 0.01.

Size distribution and characterization of vesicles shed by MDA-MB-231 cells in serum-free medium. A: Representative transmission electron micrographs of negatively stained vesicles. Bar=200 nm. B: Histogram of quantitative analysis of vesicles diameter. Mean number of analyzed vesicles per field=137. C: Proteins from whole cell lisates (WCL) and exosome-like vesicles (ELVs) were separated by electrophoresis and analysed by immunoblotting using antibodies to cytochrome c, Lysosome-associated membrane glycoprotein 1 (LAMP1) and Heat shock cognate 70 (HSC70).
Results
Purification and characterization of MDA-MB-231 derived vesicles. Electron micrographs revealed that the isolated extracellular particles consisted primarily of rounded and cup-shaped vesicles of variable size (Figure 1A). Quantitative analysis, as shown in the histogram (Figure 1B), indicate that the ELV fraction contains vesicles with a diameter ranging from 20 to 180 nm. However, the majority of the vesicles (>75%) were between 40 and 100 nm, a size which is compatible with that previously reported for ELVs (29).
In order to avoid possible contamination of our fraction by subcellular components, mainly mitochondria or apoptotic bodies, putative ELVs fraction and whole-cell lysates were then probed with specific antibodies to cytochrome c, LAMP1 and HSC70. Cytochrome c is a mitochondrial protein essential for the generation of the mitochondrial membrane potential that drives the formation of ATP, but it is also a central apoptotic effector which may remain inside the apoptotic blebs (30). LAMP1 is a highly glycosylated integral protein enriched in late endosomes and/or lysosomes of normal cells (31) and also on the cell-surface of highly metastatic tumor cells (32). HSC70 is a chaperone protein of 70 kDa, member of the HSP70 family closely associated with the synthesis, folding and secretion of proteins (33). Interestingly, LAMP1 and HSC70 proteins were observed in exosomes by several studies (24, 34). Figure 1C shows the western blotting of the three markers. Cytochrome c was exclusively detectable in the WCL and absent from the vesicle fraction. This observation indicates that the vesicle preparation was not contaminated with elements of cellular organelles or by apoptotic blebs. As expected, the vesicular markers LAMP1 and HSC70 were detected in both WCL and vesicles. This, taken together with the absence of cytochrome c in the vesicle fraction, suggests that these vesicles can be considered ELVs.
Proteomic analysis of MDA-MB-231 derived ELVs. To evaluate ELV composition in comparison with the one of the whole cell lysate, WCL and ELV proteins were separated by 2D-IPG as described under Materials and Methods. Two independent ELV preparations from MDA-MB-231 cells showed high reproducibility and data were largely overlapping (data not shown).
Representative 2D proteomic maps of WCL and ELVs are shown in Figure 2. The protein identities are marked with labels corresponding to the accession numbers of the Swiss-Prot database and the different isoforms of the same protein are jointly labelled.
One hundred and seventy-nine protein spots, corresponding to 113 genes, were identified in the maps and the identity of the proteins was assessed by N-terminal sequencing and by MALDI-MS. All the proteins identified are listed in Table I.
Quantification of spots was performed with the densitometric algorithm of the Image-Master software (Amersham), by measuring the relative abundance of 179 selected spots expressed as %Vol (integration of intensity over the spot area normalized by the sum of all the spot volumes).
For the comparative analysis of protein expression, the average of two spot values from two different gels was utilized, and expression levels of WCL and ELV fraction were considered significantly different for ≥30% variations.
Out of 179 identified proteins, 32 protein isoforms, corresponding to 22 genes, were found to be more abundant in ELVs compared to WCL. Figure 3A shows a panel of the differentially abundant protein spots, cropped from 2D gels of WCL and ELVs respectively; Figure 3B illustrates the histogram of the relative intensities of these proteins.
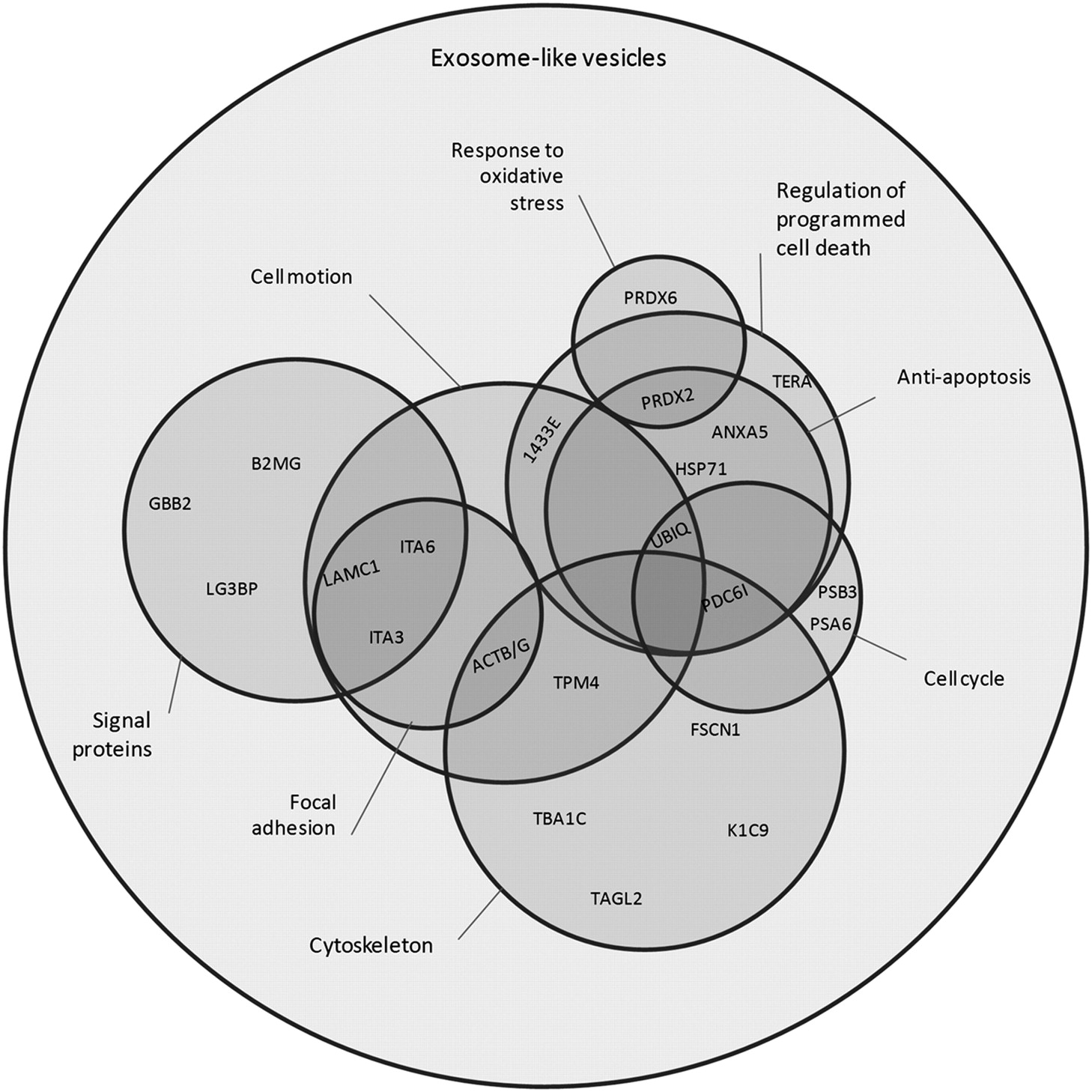
The catalogue of differentially enriched proteins in vesicles is reported in Table II. These proteins were clustered into eight functional categories, mainly according to the current bioinformatics resources. The clusters obtained are: cytoskeleton, regulation of programmed cell death and the subgroup anti-apoptosis, cell cycle, signal proteins, response to oxidative stress, focal adhesion and cell motion. In order to clarify the overlap among these clusters, a Venn diagram (Figure 4) was constructed, where the graphical partial areas are related to the number of elements within each region. Interestingly, clusters which contain a greater number of unshared proteins are the cytoskeleton, regulation of programmed cell death and signal proteins. These clusters, as reported in literature, are involved at different levels in mechanisms which have a role in the malignant progression of cancer (35-37).

Representative 2D proteomic maps of whole cell lisates (WCL) and exosome-like vesicles (ELVs). The identified proteins are indicated with the accession numbers of the Swiss-Prot database.
Catalogue of the protein spots identified in the proteomic maps of the exosome-like vesicles (ELVs). The protein names, accession numbers (AC) and abbreviated names correspond to the nomenclature used in the Swiss-Prot database. Identification methods: 1: MALDI-MS; 2: N-terminal sequencing by automated Edman degradation; 3: Western Blotting; 4: Liquid chromatography MS/MS.
The cytoskeleton cluster includes structural proteins and some accessory proteins with regulatory functions (ACTB/G, TBA1C, K1C9, two isoforms of TPM4, FSCN1, TAGL2 and two isoforms of PDC6I; see Table I), some of which may be involved in exosome biogenesis, cell cycle regulation (38), and cytoskeleton re-organization. Among the latter, transgelin-2 (TAGL2), containing a calponin-like repeat which is able to bind actin and tropomyosin, may also play a role in the shedding process. Moreover, some loss-of-function studies in si-TAGLN2 transfectants of renal carcinoma cell line showed significant inhibition of cell proliferation and invasion, indicating an oncogenic role for TAGL2 (39).
The cytoskeleton cluster also includes the programmed cell death 6-interacting protein (PDC6I), a protein greatly enriched in ELV. This protein is implicated in the concentration and the sorting of cargo proteins of the multivesicular body, which are released into the extracellular space as exosomes, and may inhibit death induced by several stimuli, through the binding to the apoptosis-linked gene 2, a Ca2+-binding protein necessary for cell death (40). Other authors demonstrated that regulators of apoptosis are characteristic of different cancer vesicles (41-44) and it is thought that cancer cells, through them, may positively or negatively influence the survival capability of surrounding cells, contributing to the formation of a favorable microenvironment (45).
The regulation of programmed cell death cluster includes the transitional endoplasmic reticulum ATPase/valosin-containing protein (TERA) and two isoforms of 14-3-3E, an adapter protein implicated in the regulation of a large spectrum of both general and specialized signaling pathways, among which cell cycle control, proliferation, apoptosis, neurogenesis and abnormal growth of tumor cells in vitro (46). Moreover, its overexpression in primary hepatocellular carcinoma tissues, in comparison with non-tumoral tissues, predicts a high risk of extrahepatic metastasis and worse survival (47). Furthermore, this protein has been also found in exosomes derived from biological fluids such as urine (48). A sub-group of the regulation of programmed cell death cluster is the anti-apoptosis cluster; this contains the proteins Annexin A5 (ANXA5) and Heat shock 70 kDa protein 1 (HSP71), which are among the major negative regulators of programmed cell death, two isoforms of Peroxiredoxin-2 (PRDX2), the Ubiquitin (UBIQ) and also PDC6I. Ubiquitin is a highly conserved nuclear and cytoplasmic protein that plays a major role in targeting cellular proteins for degradation by the 26S proteosome. It is also involved in the maintenance of chromatin structure, the regulation of gene expression, and the stress response. Ubiquitin is synthesized as a precursor protein fused to an unrelated protein, the 60S ribosomal protein L40. Ubiquitin, PDC6I, with PSB3 and PSA6 are implicated at different levels in the control of cell cycle (49).
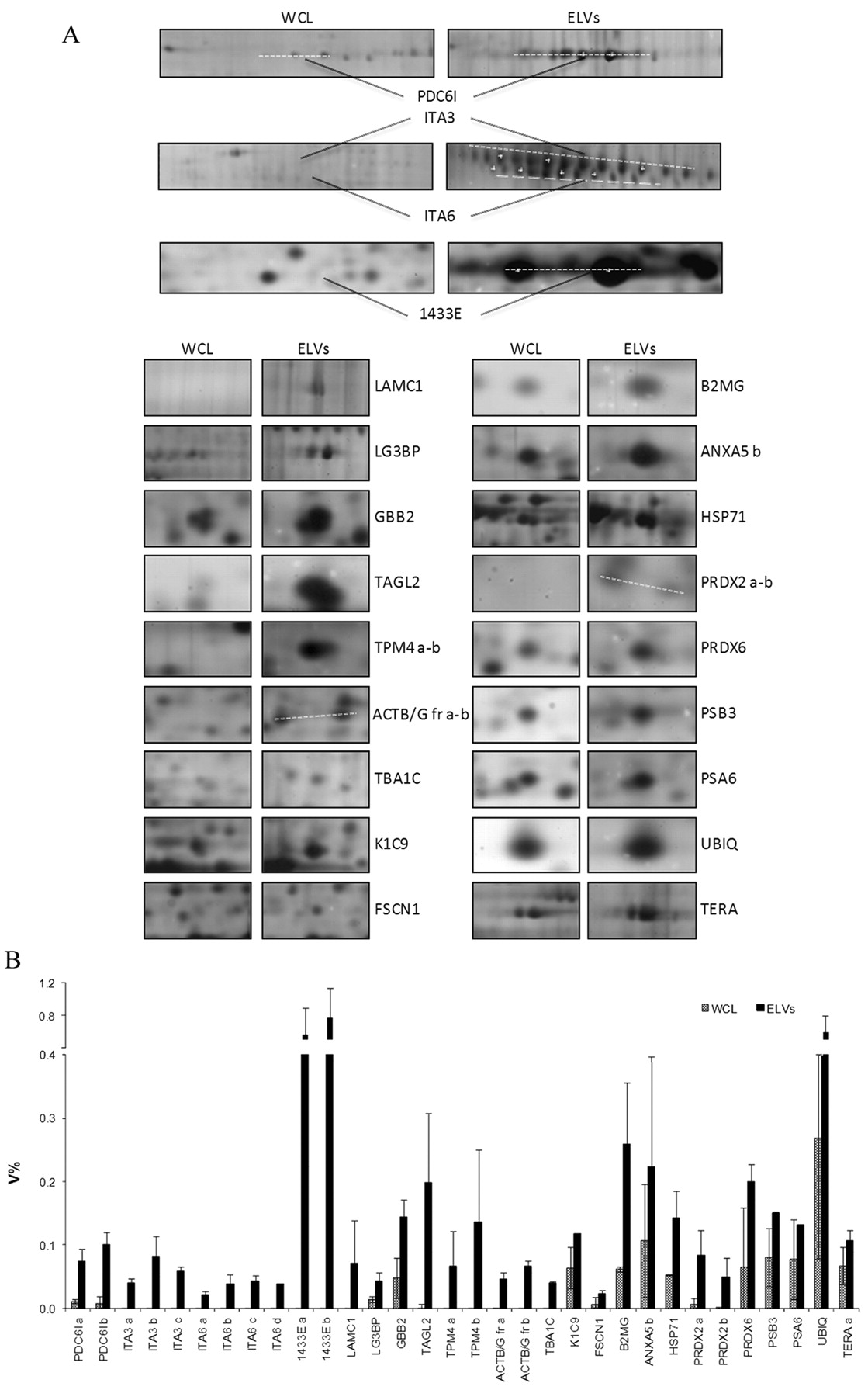
Comparative qualitative/quantitative analysis of proteins enriched in the exosome-like vesicles (ELVs) fraction in comparison with whole cell lisates (WCL). A: Panel of the differentially abundant protein spots cropped from 2D gels. B. Histogram of the relative volume (V%) of enriched proteins in the ELV fraction in comparison with WCL. For protein symbols, see Table I.
Catalogue of the protein spots enriched in the exosome-like vesicles (ELVs) fraction. The protein names, accession numbers (AC) and abbreviated names correspond to the nomenclature used in the Swiss-Prot database. Identification methods: 1: MALDI-MS; 2: N-terminal sequencing by automated Edman degradation; 3: Liquid chromatography MS/MS.
Beta-2-microglobulin (B2MG), Galectin-3-binding protein (LG3BP), Laminin subunit gamma-1 (LAMC1), Guanine nucleotide-binding protein G(I)/G(S)/G(T) subunit beta-2 (GBB2), three isoforms of Integrin alpha-3 (ITA3) and four isoforms of Integrin alpha-6 (ITA6) are grouped into the signal proteins cluster and play different roles in multiple pathways. Interestingly there is an enrichment of B2MG, the beta-chain of major histocompatibility complex class I molecules, in ELVs. Since it is involved in the presentation of peptide antigens to the immune system, it could play an important role in the immune system modulation during tumor progression (34).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Venn diagramm showing the overlap among functional clusters obtained according to the DAVID Bioinformatics Resources 6.7 (Gene Ontology Data Base).
ITA3 and ITA6 were found to be enriched in several isoforms, all grouped in to the signal proteins cluster. They are involved in cell-cell and cell-extracellular matrix adhesion and probably address exosomes to target cells in a specific manner (34).
These proteins, together with LAMC1 and ACTB/G, are also clustered as focal adhesion (50) and are a smaller part of a larger cluster, defined as cell motion that shares some proteins (1433E, UBIQ and TPM4) with other clusters.
PRDX2 and Peroxiredoxin-6 (PRDX6), proteins involved in oxidation-reduction processes, are also classified as response to oxidative stress. PRDX2 has been identified as an early-stage breast cancer auto antigen, useful in early diagnosis of aggressive breast cancer (51). PRDX6 has also been shown to be related to breast cancer malignancy; indeed its overexpression leads to a more invasive phenotype and metastatic potential (52). Interestingly, these proteins were observed in microvesicles of different origins (48, 53) but this is the first time where PRDX6 has been reported in breast cancer exosomes.
Discussion
There is a general agreement that the initial steps of carcinogenesis are sustained by genetic changes, while the progression is mainly due to epigenetic influences. One of the early stages of progression concerns the loosening and subsequent rupture of cell-cell and cell-matrix adhesion complexes. Following the detachment from the original tissues, neoplastic cells acquire a motile phenotype and display several membrane perturbations with frequent vesicle shedding, both in vivo and in vitro (27, 54, 55). As stated earlier, the extracellular vesicle population reveals some morphological typologies which have different effects on the surrounding microenvironment. Molecular transfer mediated by EVs to adjacent or remote cells has been demonstrated to influence several aspects of cell-matrix interactions, including cancer progression, thrombosis, angiogenesis and immune response modulation (8, 56-59).
The present study provides new insights into breast cancer-derived vesicles, which, according to our experimental data, may be regarded as exosome-like. This is on the basis of electron microscopical evaluation of their size and morphology, as well as through immunological identification of current vesicle markers, LAMP1 and HSC70, and by the absence of citocrome c, which testifies to the non-apoptotic origin of these vesicles. This highly purified fraction of ELVs was submitted to a systematic proteomic analysis, which revealed a distinctive protein composition that may represent the signature of breast cancer ELVs, with important roles in cancer progression. The proteins of this cluster may play a multiplicity of roles in several biological cell functions through key mechanisms attributed to the exosomes. These mechanisms include: i) direct contact between surface molecules of vesicles and cells; ii) endocytosis of vesicles; iii) vesicle-cell membrane fusion (2).
It is reasonable to assume that vesicle production allows neoplastic cells to exert different effects, according to the possible acceptor targets. For instance, vesicles could potentiate the malignant properties of neighboring neoplastic cells and/or activate non-tumoral adjacent cells, through transfer of multifunctional proteins, such as 14-3-3 epsilon or PDC6I which play roles as regulators of cell proliferation, cycle and cell death. Moreover vesicles could convey signals to immune cells and surrounding stromal cells (29). For example, some proteins, such as integrins or major histocompatibility complex, may mediate transcellular communication and immune regulations, but also induce autocrine and paracrine effects on cell motility and invasion (34, 60).
We suggest that the vesicular components of breast cancer cells are involved in tumor survival and expansion, and account for differing abilities in metastasis.
Acknowledgements
The present work was supported in part by: University of Palermo funds to M.L. Vittorelli; funds of the Operative Regional Program Sicily 3.14 (DIAMOL project) to I. Pucci-Minafra; State-made contributions to C.OB.S. (5x1000) for distinguished no-profit research associations. The Authors thank the Werner Geissberger Stiftung (Basel, Switzerland) for support to G. Palazzolo's work in Basel.
Footnotes
-
Conflict of Interest Statement
The Authors report no conflicts of interest. The Authors alone are responsible for the content and writing of this article.
- Received January 5, 2012.
- Revision received February 9, 2012.
- Accepted February 13, 2012.
- Copyright© 2012 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved