

Abstract
Background: Patients with metastatic and muscle-invasive bladder cancer are commonly treated with cisplatin. A significant proportion of patients develop disease progression after an initial response to chemotherapy. Presently there is no standard of care for such patients. We examined whether pretreatment with an epigenetic agent would result in reversal of drug resistance. Materials and Methods: Methylation of proapoptotic and cell cycle genes in bladder cancer cells was examined. Cisplatin- and docetaxel-resistant cells were generated. The effect of target of methylation-induced silencing (TMS1) expression and pretreatment of wild-type and drug-resistant cells with 5-azacytidine on chemosensitivity was determined. Results: Unidirectional crossresistance of cisplatin-resistant UMUC3 cells to docetaxel was observed. Recombinant expression of TMS1 or pre-treatment of wild-type and drug-resistant cells with 5-azacytidine resulted in enhanced sensitivity to cisplatin and docetaxel. Conclusion: Our results indicate that epigenetic therapy may restore sensitivity to chemotherapeutic agents in bladder cancer cells.
- Epigenetics
- bladder cancer
- DNA methylation
- chemotherapy
- chemoresistance
- 5-azacytidine
Bladder cancer is the fourth most diagnosed cancer among men in the United States and the eighth leading cause of cancer-related deaths, with an estimated 69,250 new cases and 14,990 deaths in 2011 (1). Approximately 25% of bladder cancer patients present with muscle invasive or metastatic disease requiring systemic chemotherapy. Although the majority of patients presenting with superficial disease have a favorable prognosis, approximately 70% of these develop disease recurrence and 10 to 20% of these patients progress to muscle-invasive bladder cancer that eventually requires systemic chemotherapy (2, 3). Cisplatin-based first-line chemotherapy regimens such as methotrexate, vinblastine, doxorubicin and cisplatin (M-VAC), dose-dense M-VAC, and gemcitabine and cisplatin (GC) have been used to treat metastatic transitional cell carcinoma (TCC) of the urothelium and have shown similar efficacy (4-6). Although these regimens have demonstrated initial high response rates of 40-70%, the median progression free and overall survival are about 8 and 15 months, respectively, indicating a highly chemoresistant disease upon relapse (7). Although bladder cancer is a chemotherapy-sensitive malignancy, a majority of patients develop disease progression after an initial response to chemotherapy. Currently, there is no standard of care in the second-line setting i.e. after progression on cisplatin-based chemotherapy. Taxanes such as docetaxel and paclitaxel have demonstrated substantial activity in bladder cancer when used as single agents in previously untreated patients (8). However, second-line study of taxanes in patients with bladder cancer have reported lower response rates than that observed for chemo-naïve patients (9, 10). Presently there are limited treatment options for patients whose disease has progressed on cisplatin- or taxane-based therapy.
The mechanisms of resistance to cisplatin and taxanes are currently unclear. Cisplatin and docetaxel induce cell death via the apoptotic pathway (11-14). Cancer cells are known to avoid apoptosis by a number of mechanisms, among which, DNA methylation-induced transcriptional repression of genes involved in the apoptotic pathway plays an important role (15-17). Activation of these genes by epigenetic therapy could lead to novel therapeutic approaches in bladder cancer.
Target for methylation-induced silencing (TMS1)/apoptosis-associated speck-like protein containing caspase recruitment domain (CARD) (ASC) is characterized by the presence of pyrine domain (PYD) and CARD motifs and has been shown to induce apoptosis in a caspase-8 dependent manner. It can also modulate the activity of caspase-1 and block the downstream activation of nuclear factor kappa B (18). The role of TMS1/ASC in apoptosis is of interest as studies have shown that the gene is epigenetically silenced by methylation in many tumor types and thus may have an important role in chemoresistance, tumorigenesis, and progression of the disease (19). Aberrant methylation of TMS1 also correlated with gene silencing in 70% of small cell lung cancer cases (20). Methylation-mediated silencing of TMS1 was also observed in ovarian cancer cell lines and correlated with a survival advantage of tumor cells, indicating TMS1 may contribute to ovarian tumorigenesis (21). It has been shown that overexpression of TMS1 induces apoptosis and inhibits growth of breast cancer cells (22).
Primers for MS-PCR analysis.
Since methylation-mediated silencing of genes involved in apoptotic pathway can contribute to chemoresistance, we examined whether epigenetic therapy can reverse chemoresistance in bladder cancer cells.
Materials and Methods
Cell culture. Bladder cancer cells were purchased from the American Type Culture Collection (ATCC), Manassas, VA, USA. UMUC3, T24 and TCCSUP bladder cancer cells were routinely cultured in RPMI-1640 medium (Mediatech, Manassas, VA, USA) supplemented with 10% fetal bovine serum, 2 mM glutamine (Invitrogen, Carlsbad, CA, USA) and 100 μg/ml penicillin-streptomycin (Invitrogen) in a humidified incubator at 37°C with 5% CO2. Docetaxel and cisplatin were procured from LC Laboratories (Woburn, MA, USA) and Sigma Aldrich (St. Louis, MO, USA) respectively. 5-azacytidine was provided by Celgene Corporation.
Drug-resistant cells. UMUC3 cells resistant to 5 nM docetaxel (UMUC3-5DR) or 1 μM cisplatin (UMUC3-1CR) were selected by culturing cells in docetaxel or cisplatin respectively in a dose-escalating manner. After cells sensitive to drug were no longer present and the surviving UMUC3 cells had repopulated the flask and continued to divide through four passages, the concentration of drug in the medium was increased. This was continued in a stepwise manner until final concentrations of 5 nM docetaxel or 1 μM cisplatin were reached. UMUC3-5DR and UMUC3-1CR cells were maintained in medium containing 5 nM docetaxel and 1 μM cisplatin respectively. Using a similar strategy, T24 cells resistant to 20 nM docetaxel (T24-20DR) and TCCSUP cells resistant to 10 nM docetaxel (TCCSUP-10DR) were also generated.
Transfection. UMUC3 cells were transfected with either pCMV6-XL5/TMS1 plasmid or pCMV6-XL5/Mock plasmid (Origene, Rockville, MD, USA). Transfection was carried out using Amaxa Nucleofection system (Lonza, Walkersville, MD). Cells were then seeded in 96-well plates and treated with either cisplatin or docetaxel for 72 h. Cell viability was measured after 24, 48 and 72 h using Cell Titer Blue (Promega, Madison, WI, USA).
Drug treatment. Wild-type and drug resistant bladder cancer cells were treated with different concentrations of docetaxel/cisplatin and cell viability was measured 72 h following treatment using Cell Titer Blue. Cells were treated with different concentrations of 5-azacytidine for 72 h, after which RNA/DNA was extracted. For combination treatment, wild-type and drug-resistant cells were seeded in 96-well plates and treated with 5-azacytidine for 72 h followed by treatment with docetaxel or cisplatin for 72 h. Cell viability was measured after 24, 48 and 72 h using Cell Titer Blue.
Cell viability assay. Cells were incubated with RPM1 medium containing Cell Titer Blue for 5 h and fluorescence (560Ex/590Em) was measured in a Biotek Synergy HT multi-task plate reader.
Methylation-specific polymerase chain reaction (MS-PCR). Methylation patterns of Reprimo, Dicer1 (DCR1), ras association domain family 1 (RASSF1A), target of methylation induced silencing (TMS1), PDZ and LIM domain protein 4 (PDLIM4) and E-cadherin (ECAD) in the three bladder cell lines were analyzed by MS-PCR. These genes have been previously shown to play a role in apoptosis and/or cell cycle regulation (23-25). DNA was extracted using the Masterpure DNA purification kit (Epicentre, Madison, WI, USA) as per manufacturer's instructions. Bisulfite conversion of genomic DNA was carried out as previously described (26, 27) by incubating 5 μg DNA with a 5 M bisulfite solution and 100 mM hydroquinone, pH 5.0 at 55°C for 4 h. This was followed by desulfonation by addition of 3 M NaOH, and desalting using a QIAquick PCR purification kit (Qiagen, Valecia, CA, USA). After bisulfite conversion, the DNA template was amplified with primers sets specific for the methylated gene promoter sequence (Table I). The PCR products were then analyzed by agarose gel electrophoresis and visualized under UV light.
RNA extraction and quantitative reverse transcriptase PCR (RT-PCR). RNA was extracted from cells using the Masterpure RNA purification kit (Epicentre) as per the manufacturer's instructions and reverse transcribed using MMLV Reverse Transcriptase (USB Corporation, Cleaveland, OH). Real time PCR amplification was performed in triplicates with cDNA using primers for TMS1. The average Ct was then used to quantitate relative mRNA levels by the comparative Ct method. The levels of TMS1 were normalized to that of the housekeeping gene, glyceraldehyde 3-phosphate dehydrogenase (GAPDH). The primer sequences used to amplify TMS1 were: forward: 5’ GCA GCC AAG CCA GGC C 3’and reverse: 5’ CCA GCA GCC ACT CAA CGT T 3’ and the sequences used to amplify GAPDH were forward: 5’ GGT ATC GTG GAA GGA CTC ATG AC 3’ and reverse: 5’ CAC GCC ACA GTT TCC CGG A 3’. The PCR reaction was carried out in a volume of 25 μl using iQ™ SYBR® Green Supermix in a MyiQ™ Single-Color Real-Time PCR Detection System (BioRad, Hercules, CA, USA).
Results
Methylation status of genes involved in apoptosis and/or cell cycle regulation in bladder cancer cells. Using MS-PCR, we examined the methylation status of Reprimo, DCR1, RASSF1A, TMS1, PDLIM4 and ECAD. Reprimo, DCR1, RASSF1A and ECAD were found to be methylated in all three cell lines. TMS1 was methylated in TCCSUP and UMUC3 but not in T24 cells. PDLIM4 was methylated in T24 and TCCSUP cells and unmethylated in UMUC3 cells (Table II).
Reactivation of TMS1 by treatment with 5-azacytidine. We have previously shown that TMS1 contributes to chemosensitivity in breast and pancreatic cancer cells (15, 17). Since TMS1 was found to be methylated in UMUC3 cells, we examined the role of TMS1 in chemosensitivity in these cells. To confirm the role of DNA methylation of the TMS1 promoter in regulation of gene expression, UMUC3 cells were treated with 0.6 μM 5-azacytidine for 72 h after which promoter methylation and gene expression levels were determined. Treatment with 5-azacytidine resulted in demethylation of the TMS1 promoter (Figure 1A) concomitant with re-expression of TMS1 (Figure 1B).
Overexpresson of TMS1 results in enhanced sensitivity of UMUC3 cells to cisplatin and docetaxel treatment. The role of TMS1 in apoptosis is well documented (18, 22, 28-32). However, its role in sensitivity to cisplatin and docetaxel has not been well understood. We determined the effect of recombinant expression of TMS1 in UMUC3 cells on sensitivity to cisplatin and docetaxel. UMUC3 cells were transfected with TMS1 expression vector or empty vector (mock) and treated with either cisplatin or docetaxel for 72 hours after which cell viability was measured. Cells transfected with TMS1 expression vector showed increased sensitivity to both cisplatin and docetaxel treatment when compared to mock-transfected cells. Increased cell death was observed in UMUC3 cells expressing TMS1 as compared to mock-transfected cells when treated with 1.25 μM cisplatin (47% vs. 23%) and with 2.5 μM cisplatin (61% vs. 45%) (Figure 1C). Increased cytotoxicity was observed in UMUC3 cells expressing TMS1 as compared to mock-transfected cells when treated with 1.25 nM docetaxel (56% vs. 33%) and with 2.5 nM docetaxel (61% vs. 42%) (Figure 1D).
Methylation status of genes related to apoptosis and cell cycle regulation in bladder cancer cell lines used in this study.
Pretreatment with 5-azacytidine results in enhanced cytotoxicity to cisplatin and docetaxel treatment in UMUC3 cells. Our previous studies in prostate, breast cancer and pancreatic cells have shown that combined treatment with 5-azacytidine and chemotherapeutic drug improves the cytotoxic effect of chemotherapeutic drugs (15-17). We hypothesized that induction of expression of TMS1 and other genes involved in apoptosis and cell cycle regulation by 5-azacytidine pretreatment would increase sensitivity of UMUC3 cells to cisplatin and docetaxel chemotherapy. Cells were treated with concentrations of 5-azacytidine ranging from 0.3 μM to 5 μM and cell survival was measured after 24, 48, and 72 h (data not shown). Quantitative RT-PCR was used to measure TMS1 gene expression after 72 h. Treatment with 0.6 μM 5-azacytidine resulted in 75% viable cells after 72 h and ~150-fold increase in TMS1 mRNA levels. Hence, we chose the sub-cytotoxic dose of 0.6 μM 5-azacytidine for pretreatment of UMUC3 cells.
UMUC3 cells were pre-treated with 0.6 μM 5-azacytidine for 72 h followed by treatment with either cisplatin or docetaxel for 72 h, following which cell viability was measured. Pretreatment with 5-azacytidine resulted in increased cytotoxicity of both cisplatin and docetaxel. UMUC3 cells pretreated with 5-azacytidine showed a 64% increase in cell death when treated with 1 μM cisplatin compared to cells with no pretreatment (Figure 2A). Similarly, pretreatment of cells with 5-azacytidine increased cytotoxicity of 5 nM docetaxel by 30% compared to cells that were treated with docetaxel alone (Figure 2B).
Characterization of drug resistant UMUC3 cells. In order to confirm resistance of UMUC3-1CR and UMUC3-5DR cells to cisplatin and docetaxel respectively, UMUC3 wild-type and drug-resistant cells were treated with different concentrations of cisplatin and docetaxel for 72 h. UMUC3-1CR cells showed resistance to cisplatin compared to UMUC3 wild-type cells (2% vs. 23% cytotoxicity). UMUC3-5DR cells also showed resistance to docetaxel compared to UMUC3 wild-type cells (2% vs. 72% cytotoxicity) (data not shown).

A: MSPCR analysis. UMUC3 cells were treated with 0.6 μM 5-azacytidine (Aza) for 72 h. Control cells were left untreated. Genomic DNA was extracted from cells, bisulfite treated and analyzed by MS-PCR using primers specific for unmethylated (U) and methylated (M) sequences. B: Gene epression analysis. RNA extracted from cells was reverse transcribed and amplified by quantitative RT-PCR. TMS1 expression levels were normalized to that of the housekeeping gene, GAPDH. C, D: Effect of TMS1 expression on sensitivity to chemotherapeutic agents. UMUC3 cells were transfected with TMS1 expression vector or vector alone (mock) using Amaxa nucleofection kit and seeded in 96-well plates for 24 h, following which they were treated with cisplatin (Cis) (C) or docetaxel (Doc) (D) for 72 h. Cell viability was assayed by cell-titer blue assay.
Reversal of drug resistance in UMUC3 cells by pretreatment with 5-azacytidine. UMUC3-1CR and UMUC3-5DR cells were pre-treated with 5-azacytidine for 72 h followed by treatment with cisplatin and docetaxel respectively for 72 h, following which cell viability was measured. Pretreatment with 5-azacytidine resulted in increased cytotoxicity of both cisplatin and docetaxel. UMUC3-1CR cells pretreated with 5-azacytidine showed a 44% increase in cell death when treated with 1 μM cisplatin compared to cells with no pretreatment (Figure 2C). Similarly, UMUC3-5DR cells pretreated with 5-azacytidine showed an increase in cytotoxicity of 5 nM docetaxel by 55% compared to cells that were treated with docetaxel alone (Figure 2D).
Crossresistance between cisplatin and docetaxel in UMUC3 cells. Docetaxel is used to treat bladder cancer patients who are resistant to cisplatin chemotherapy (9). Epigenetic silencing of genes contributing to resistance to a chemotherapeutic agent may also affect sensitivity to other chemotherapeutic drugs. Determining the sensitivity to docetaxel in cisplatin resistant cells and vice versa may improve our knowledge on the interactions of these drugs and aid in the development of novel therapeutic regimens for drug resistant bladder cancer. To determine whether UMUC3 cells show cross resistance between cisplatin and docetaxel, we treated UMUC3-1CR cells with different concentrations of docetaxel. UMUC3-1CR cells showed reduced sensitivity to docetaxel compared to wild type cells (Figure 3A). However, UMUC3-5DR cells did not show much difference in sensitivity to cisplatin compared to wild-type cells (Figure 3B). Furthermore, we examined the effect of combination of 5-azacytidine and docetaxel in UMUC3-1CR cells. UMUC3-1CR cells pretreated with 5-azacytidine showed a 32% increase in cell death when treated with 5 nM docetaxel compared to cells with no pretreatment (Figure 3C). Similarly, we determined the efficacy of combination of 5-azacytidine and cisplatin in UMUC3-5DR cells. UMUC3-5DR cells pretreated with 5-azacytidine showed a 58% increase in cell death when treated with 1 μM cisplatin compared to cells with no pretreatment (Figure 3D).
Increased sensitivity of UMUC3 cells to cisplatin (A) and docetaxel (B) by pretreatment with 5-azacytidine. Cells seeded in 96-well plates were treated with 0.6 μM 5-azacytidine (Aza) for 72 h following which they were treated with cisplatin (Cis) or docetaxel (Doc) for 72 h. In parallel, control cells were left untreated for 72 h, following which they were treated with cisplatin or docetaxel for 72 h. Cell viability was assayed by cell-titer blue assay. Reversal of resistance of UMUC3 cells to cisplatin (C) and docetaxel (D) by pretreatment with 5-azacytidine. UMUC3 cells resistant to 1 μM cisplatin (UMUC3-1CR) and cells resistant to 5 nM docetaxel (UMUC3-5DR) were seeded in 96-well plates and treated with 0.6 μM 5-azacytidine (Aza) for 72 h, following which they were treated with cisplatin (Cis) and docetaxel (Doc) respectively for 72 h. In parallel, control cells were left untreated for 72 h, following which they were treated with cisplatin or docetaxel for 72 h. Cell viability was assayed by cell-titer blue assay.
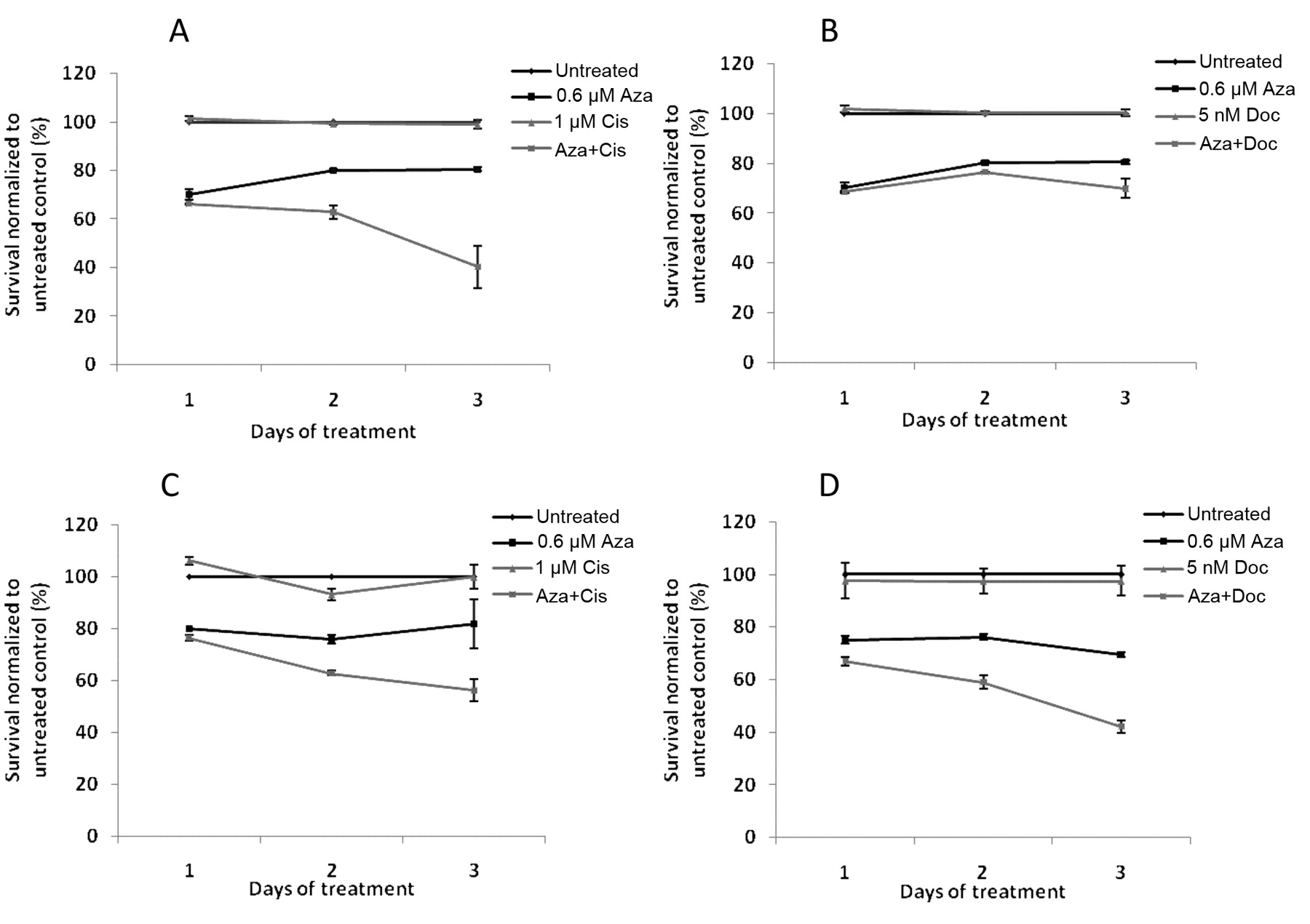
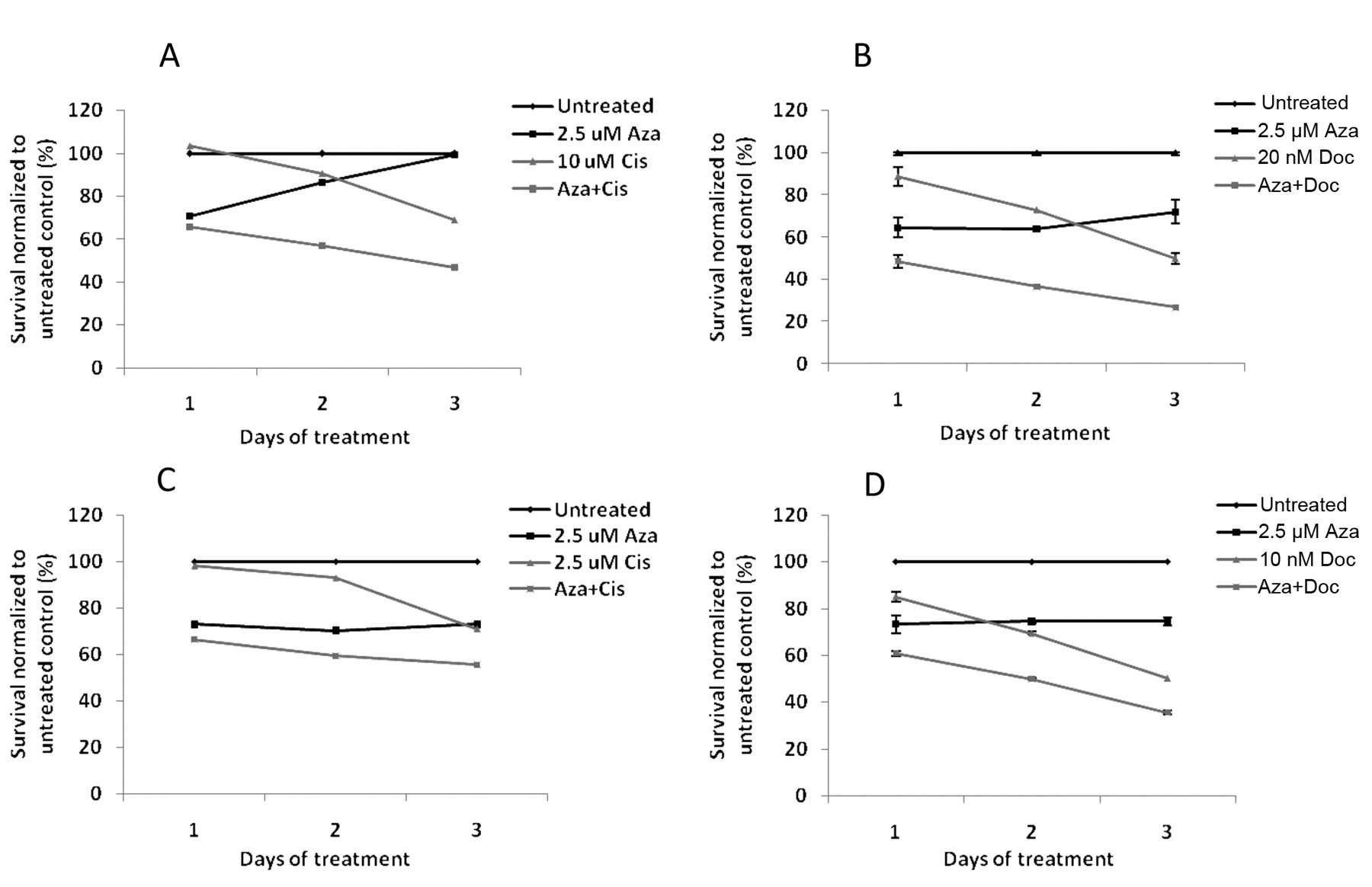
Pretreatment with 5-azacytidine results in enhanced cytotoxicity to cisplatin and docetaxel treatment of T24 and TCCSUP cells. Based on dose titration on T24 and TCCSUP cells (data not shown), we chose the sub-cytotoxic dose of 2.5 μM of 5-azacytidine for pretreatment. T24 and TCCSUP cells were pre-treated with 5-azacytidine for 72 h followed by treatment with either cisplatin or docetaxel for 72 h, following which cell viability was measured. Pretreatment of cells with 5-azacytidine resulted in increased cytotoxicity of both cisplatin and docetaxel. T24 cells pretreated with 5-azacytidine showed a 22% increase in cell death when treated with 10 μM cisplatin compared to cells with no pretreatment (Figure 4A). Similarly, pretreatment with 5-azacytidine increased cytotoxicity of 20 nM docetaxel by 23% compared to cells that were treated with docetaxel alone (Figure 4B). TCCSUP cells pretreated with 5-azacytidine showed a 15% increase in cell death when treated with 2.5 μM cisplatin compared to cells with no pretreatment (Figure 4C) and pre-treatment with 5-azacytidine increased cytotoxicity of 10 nM docetaxel by 14% compared to cells that were treated with docetaxel alone (Figure 4D).

Crossresistance of drug-resistant UMUC3 cells. UMUC3-1CR cells were seeded in 96-well plates for 24 h, following which they were treated with different concentrations of docetaxel. Cell viability was measured 72 h post drug treatment using cell titer blue assay (A). Similarly, sensitivity of UMUC3-5DR cells to cisplatin was determined (B). Increased sensitivity of UMUC3-1CR and UMUC3-5DR cells to docetaxel (C) and cisplatin (D) by pretreatment with 5-azacytidine. UMUC3 cells resistant to 1 μM cisplatin (UMUC3-1CR) and cells resistant to 5 nM docetaxel (UMUC3-5DR) were seeded in 96-well plates and treated with 0.6 μM 5-azacytidine (Aza) for 72 h, following which they were treated with docetaxel (Doc) and cisplatin (Cis) respectively for 72 h. In parallel, control cells were left untreated for 72 h, following which they were treated with docetaxel or cisplatin for 72 h. Cell viability was assayed by cell-titer blue assay. wt: wild-type cells.
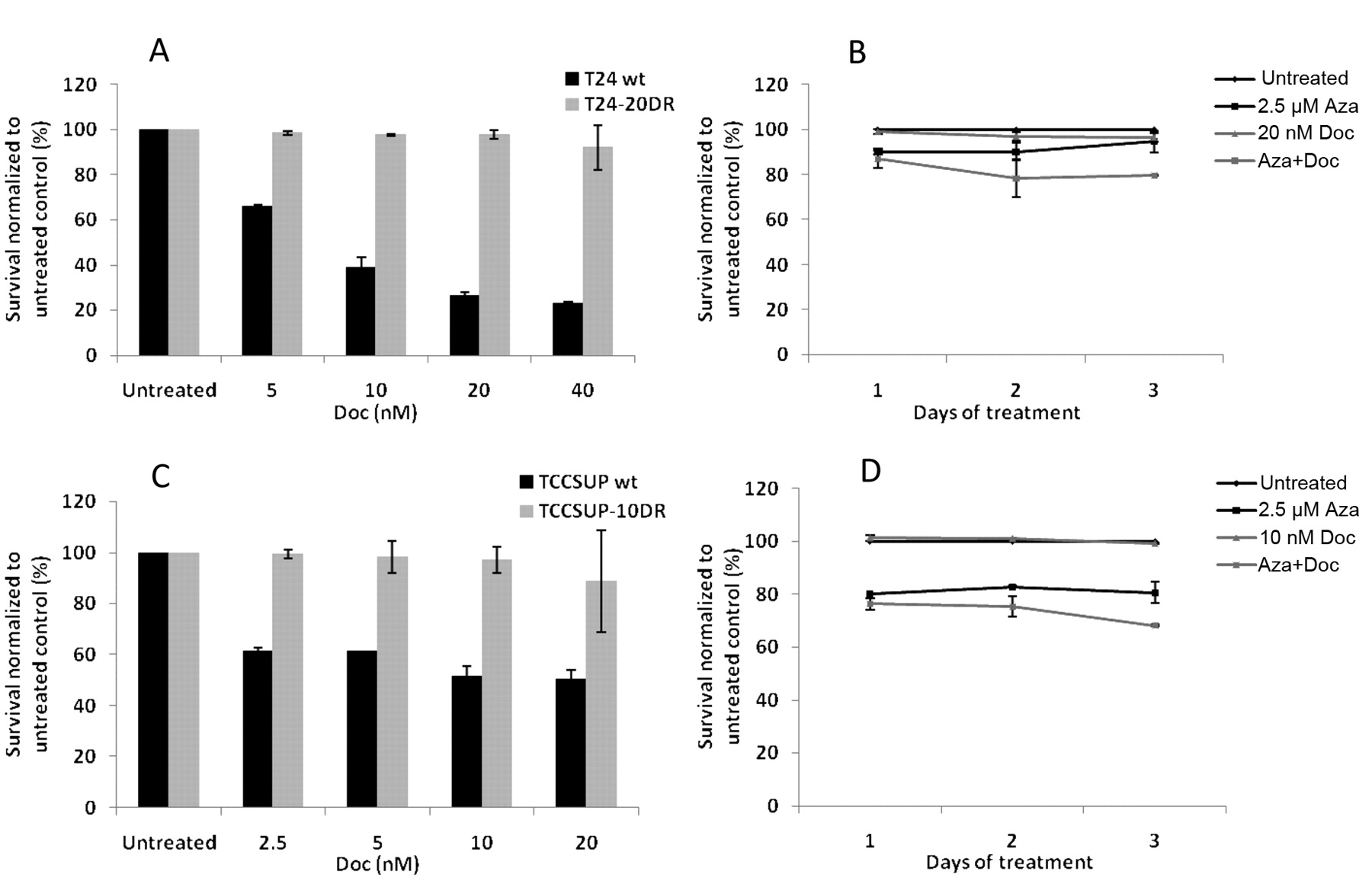
Characterization of docetaxel resistant T24 and TCCSUP cells and reversal of drug resistance by pretreatment with 5-azacytidine. In order to confirm resistance of T24-20DR cells and TCCSUP-10DR to docetaxel respectively, wild-type and drug-resistant cells were treated with different concentrations of docetaxel for 72 h. As seen in Figure 5A, T24-20DR cells showed resistance to docetaxel compared to T24 wild-type cells (2% vs. 74% cytotoxicity). TCCSUP-10DR cells also showed resistance to docetaxel compared to TCCSUP wild-type cells (3% vs. 48% cytotoxicity) (Figure 5C). T24-20DR and TCCSUP-10DR cells were pretreated with 2.5 μM 5-azacytidine for 72 h followed by treatment with docetaxel respectively for 72 h, following which cell viability was measured. Pretreatment with 5-azacytidine resulted in increased cytotoxicity of docetaxel. T24-20DR cells pretreated with 5-azacytidine showed a 17% increase in cell death when treated with 20 nM docetaxel compared to cells with no pretreatment (Figure 5B). Similarly, pre-treatment of TCCSUP-10DR cells with 5-azacytidine increased cytotoxicity of 10 nM docetaxel by 31% compared to cells that were treated with docetaxel alone (Figure 5D).
Discussion
The key finding of this study is that pretreatment of bladder cancer cells with 5-azacytidine enhances sensitivity to cisplatin and docetaxel treatment. These results suggest that up-regulation of epigenetically silenced genes results in increased cell death after the combination treatment. There is increasing evidence that epigenetic silencing is a common mechanism by which cancer cells inactivate tumor suppressor and pro-apoptotic genes. These genes are essential for apoptosis induced in response to treatment with chemotherapeutic agents (33). One of the early pieces of evidence for the role of DNA methylation in disruption of the apoptotic pathway was obtained in malignant melanoma cells in which apoptotic protease activating factor 1 (APAF-1) is silenced by DNA methylation. The authors showed that treatment with DNA methyl transferase (DNMT) inhibitor results in reactivation of the gene (34). This suggested the potential clinical benefits of reversing epigenetic silencing leading to reactivation of key genes necessary for response to chemotherapy. Over the years, a number of epigenetically silenced pro-apoptotic and tumor suppressor genes have been identified in cancer. Friedrich et al. have reported increased methylation of promoter regions of the apoptosis-related genes death associated protein kinase (DAPK), B-cell lymphoma 2 (BCL2), telomerase reverse transcriptase (TERT), RASSFIA, and tumor necrosis factor receptor superfamily, member 25 (TNFRSF25) in bladder cancer tissue compared with nonmalignant adjacent tissue. Furthermore, methylation levels of RASSF1A correlated with tumor stage in bladder cancer patients (35). Maruyama et al. have shown that RASSFIA is methylated in 35% of bladder tumors (36). Aberrant promoter methylation of Reprimo was observed in 19% of bladder tumors and was rarely detected in non-malignant tissues (37). Methylation frequency of ECAD was 36% in bladder cancer and ECAD methylation was associated with poor survival in these patients (36). DCR1 was methylated in 22% of bladder cancer tissues (38). Although previous reports showed low frequency of TMS1 methylation in bladder cancer (3.6%) (39), our results indicate a potential role for TMS1 in cisplatin and docetaxel chemotherapy of UMUC3 bladder cancer cells. In a previous study on breast cancer, we identified the potential role of TMS1 in sensitivity to docetaxel chemotherapy. TMS1 was found to be down regulated by promoter methylation in the breast cancer cell line SKBR3. Pre-treatment with a DNMT inhibitor, 5-azacytidine, increased the sensitivity of these cells to docetaxel, likely due to the up-regulation of TMS1 (15). We have also shown that recombinant expression of TMS1 in MIAPaCa-2 pancreatic cancer cells enhances sensitivity to docetaxel and gemcitabine suggesting the role of TMS1 in apoptosis induced by docetaxel and gemcitabine. In a prior study, we described methylation-mediated silencing of growth arrest and DNA damage inducible alpha (GADD45α), a gene involved in the apoptotic pathway and cell cycle control, in DU145 prostate cancer cells. Up-regulation of GADD45α, either by recombinant gene expression or by treatment with 5-azacytidine, resulted in enhanced sensitivity to docetaxel treatment (16). Based on these findings, we are currently conducting a phase I/II clinical trial using a combination of 5-azacytidine, docetaxel and prednisone in patients with docetaxel-refractory metastatic castration-resistant prostate cancer (NCT00503984).

Increased sensitivity of T24 and TCCSUP cells to cisplatin and docetaxel by pretreatment with 5-azacytidine. T24 (A, B) and TCCSUP (C, D) cells seeded in 96-well plates were treated with 0.6 μM 5-azacytidine (Aza) for 72 h, following which they were treated with cisplatin (Cis) (A, C) or docetaxel (Doc) (B, D) for 72 h. In parallel, control cells were left untreated for 72 h, following which they were treated with cisplatin or docetaxel for 72 h. Cell viability was assayed by cell-titer blue assay.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
A: Sensitivity of T24 wild-type and docetaxel-resistant T24 cells to docetaxel. T24 wild-type (wt) and T24 cells resistant to 20nM docetaxel (T24-20DR) were seeded in 96-well plates and were then treated with different concentrations of docetaxel (Doc) for 72 h. Control cells were left untreated for 72 h. B: Increased sensitivity of T24-20DR cells to docetaxel by pretreatment with 5-azacytidine. T24-20DR cells seeded in 96-well plates were treated with 0.6 μM 5-azacytidine (Aza) for 72 h, following which they were treated with docetaxel (Doc) for 72 h. In parallel, control cells were left untreated for 72 h, following which they were treated with docetaxel for 72 h. Cell viability was assayed by cell-titer blue assay. Similarly, the sensitivity of TCCSUP wild-type and docetaxel-resistant cells to docetaxel was determined (C). Effect of pretreatment of TCCSUP-10DR cells with 5-azacytidine on docetaxel sensitivity was also determined (D).
We have established cisplatin- and docetaxel-resistant bladder cancer cell lines and observed that pretreatment with 5-azacytidine reversed chemoresistance in these cells. Interestingly, we observed increased cytotoxicity when cisplatin-resistant UMUC3 cells were treated with a combination of 5-azacytidine and docetaxel and vice versa. These results suggest that a combination of 5-azacytidine and docetaxel may be used to treat cisplatin-refractory bladder cancer and bladder cancer that is refractory to docetaxel may be sensitive to combination of 5-azacytidine and cisplatin. We observed crossresistance between cisplatin and docetaxel in resistant cells. Interestingly, this crossresistance was unidirectional: cisplatin-resistant cells were resistant to docetaxel but docetaxel-resistant cells were sensitive to cisplatin. This result is consistent with the previous report by Pu et al. who showed that although cisplatin-resistant bladder cancer cells harbor crossresistance to paclitaxel, paclitaxel-resistant cells are sensitive to cisplatin (40). Presently, the mechanism of crossresistance between cisplatin and docetaxel is unclear. One study found that pre-treatment with paclitaxel of cisplatin-resistant cell lines increased the accumulation of cisplatin and was associated with tubulin alterations (41, 42). It is possible that docetaxel-resistant bladder cancer cells have similar tubulin alterations as a result of which they may accumulate more cisplatin and become sensitive to cisplatin treatment. Furthermore, we found that pretreatment of cisplatin-resistant cells with 5-azacytidine enhanced the sensitivity of these cells to docetaxel treatment, indicating that epigenetic silencing of genes may contribute to crossresistance as well.
Our results indicate that treatment with a combination of 5-azacytidine and docetaxel may be a better option for cisplatin-resistant bladder cancer compared to second-line treatment with single-agent docetaxel. Moreover, these chemoresistant cells may serve as models for elucidating mechanisms of resistance to chemotherapy in bladder cancer. Currently, the mechanisms of chemoresistance are unclear. There are presently very few reports in literature that have described the pathways that are selectively disrupted by DNA methylation during the development of chemoresistance in cancer cells. Li et al. (43) examined genome-wide gene expression and methylation changes observed in cisplatin-sensitive and cisplatin-resistant ovarian cancer cell lines and discovered novel and previously reported pathways and gene ontology groups that likely mediate cisplatin resistance. These pathways affected by epigenetic silencing included cell cycle progression, response to DNA damage and various metabolic processes in the cell. Interestingly, the authors reported a positive association between the total number of hypermethylated CpGs and increased drug resistance during the progression of cisplatin resistance (43).
In summary, the results of our study indicate that epigenetic therapy may facilitate restoration of sensitivity to chemotherapeutic agents in bladder cancer. Studies to understand the mechanisms underlying chemoresistance are presently being performed.
Acknowledgements
This work was supported by funding from Celgene Corporation to RS.
- Received August 15, 2011.
- Revision received October 7, 2011.
- Accepted October 10, 2011.
- Copyright© 2011 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved