

Abstract
Background: Chimeric T-cell antigen receptors (CAR) provide a promising approach for adoptive T-cell immunotherapy of cancer. Extensive studies on CARs have been conducted, but the detailed molecular mechanisms of the activation of a CAR-grafted T-cell remain ambiguous. This study constructed a CAR bearing anti-carcinoembryonic antigen (CEA) derived from a human monoclonal antibody (clone C2-45), and investigated the molecular basis of the CAR-mediated activation in Jurkat T-cells. Materials and Methods: A gene of a single chain fragment variable (scFv) specific for CEA was functionally cloned by the phage display method. The scFv gene was fused to human cDNAs coding for transmembrane and cytoplasmic domains of CD28 and an intracellular domain of CD3ζ. The resultant CAR45-28ζ was transiently expressed in Jurkat cells, and T-cell activation was examined by Western blotting and a cytokine production assay. A fluorescent protein-tagged ZAP-70 was used to determine whether CAR45-28ζ and ZAP-70 were co-localized at the cell surface by confocal microscopy. Results: A Western blot analysis showed CAR45-28ζ activated the ERK JNK, and p38 pathways in a CEA-dependent manner. An immunofluorescent analysis revealed the CEA-dependent formation of the signaling clusters at the antigen-CAR interface. Conclusion: CAR45-28ζ induced a wild-type T-cell receptor-like molecular event upon CEA binding, suggesting that this CAR fused gene may be useful for cancer therapy.
- Carcinoembryonic antigen
- CEA
- chimeric T-cell antigen receptor
- CAR
- human antibody
- signaling cluster
- T-cell activation
Adoptive immunotherapy involving the transfer of lymphokine-activated killer (LAK) cells or tumor infiltrating lymphocytes (TILs) has attracted significant attention (1, 2). However, the clinical application of these therapies has not necessarily worked well due to the weak trafficking ability of LAK cells to the tumor site and difficulties in isolating the effector populations and their expansion with high-dose interleukin (IL) -2 (3, 4). In addition, the effectiveness of such therapies and other HLA-dependent therapy, such as cancer vaccination, cannot always be expected, since HLA-peptide complexes are frequently lost or down-regulated in many types of human tumors (5, 6).
Chimeric T-cell antigen receptor (CAR) provides a promising approach for adoptive T-cell immunotherapy for cancer, especially in the context of tumor cells that fail to express HLA-peptide complexes and co-stimulatory molecules. CAR is typically composed of a single chain fragment variable (scFv) of the antibody specific for the tumor-associated antigens (TAAs) linked through a hinge region to transmembrane and cytoplasmic domains of T-cell signaling molecules, such as CD3ζ and CD28 (7, 8). There are obvious limitations to their clinical application, mostly due to the xenogenic origin of the scFvs, which might trigger a host immune response. A majority of the CARs reported so far have contained murine-derived or ‘humanized’ scFv for specific recognition of TAA, which could be immunogenic in human (9). Notably, extensive studies on therapeutic application of CARs have been performed, but the detailed molecular mechanisms of the CAR-mediated activation of the genetically modified T-cells are still unclear.
Our previous report described the establishment of a hybridoma clone that produces fully human monoclonal antibody specific for the carcinoembryonic antigen (CEA), one of the most important TAAs, using the KM™ mouse that carries human immunoglobulin genes instead of the endogenous immunoglobulin genes (10). The scFv gene was cloned from a human anti-CEA-producing hybridoma C2-45 and CARs were constructed using only fully human genes. CAR-transfected Jurkat cells were examined by immunoblotting and confocal immunofluorescence microscopy after stimulation with CEA, in order to investigate the CAR-mediated T-cell activation mechanisms, signaling pathways and molecular localization.

Schematic representations of CAR45-ζ and CAR45-28ζ. VH, variable region of heavy chain; VL, variable region of light chain; TM, transmembrane domain; Cyto, cytoplasmic domain.
Materials and Methods
cDNA cloning of anti-CEA scFv. mRNA was extracted from the hybridoma clone C2-45 with the QuickPrep mRNA purification kit (GE Healthcare, Chalfont St. Giles, UK). The cDNAs coding variable regions of VH and VL genes were PCR amplified using a mixture of the human V-gene specific forward and backward primers (as described in ref 11, with minor modifications) using the One-Step reverse transcription PCR (RT-PCR) kit (Stratagene, La Jolla, CA). An scFv linker DNA that connects VH to VL was prepared by PCR amplification using modified RHuJH and RHuVκ primers and phagemid vector pIT2 containing anti-ubiquitin scFv as a template (11, 12). The VH and VL fragments were assembled into the scFv form by splice overlap extension PCR. The PCR products were ligated into the SfiI and NotI sites of pIT2. The resulting plasmid was transformed into Escherichia coli TG-1. Phage particles displaying anti-CEA scFv were prepared using KM13 helper phage as described elsewhere (13), and an affinity panning was performed against a CEA-coated Maxisorp immunotube (Nunc, Roskilde, Denmark). Phage binders were eluted with trypsin-phosphate buffered saline (PBS). The eluted phages were infected into TG-1 cells, and then they were plated out on 2×TY plate containing 100 μg/ml ampicillin. The colonies were picked up, and the monoclonal phage was prepared from each isolate. The binders specific for CEA were selected by phage ELISA using HRP-conjugated anti-M13 phage antibody (GE Healthcare) with TMB One Solution (Promega, Madison, WI). Plasmid was prepared from selected clone and sequenced with the Model 3100 DNA sequencer (Applied Biosystems, Foster City, CA) using the BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems).
Construction of CAR45-ζ and CAR45-28ζ genes. Each Flexi®ORF clone ORS10195 and ORS10193 (Promega) was used as a template DNA for PCR amplification of the leader sequence and hinge region cDNAs of human CD8, and the transmembrane and/or cytoplasmic domains of CD3ζ. The cDNA coding for the transmembrane and cytoplasmic domains of human CD28 were amplified by PCR from the CD28 cDNA as described elsewhere (14). The cDNA of the anti-CEA scFv was reamplified to append EcoRI and HindIII sites at the 5′ and 3′ ends respectively. These gene fragments were genetically linked together step-by-step, while constructing two forms of CAR, namely CAR45-ζ and CAR45-28ζ, as shown in Figure 1. The resulting CAR genes were cloned into the pBI-CMV4 mammalian bicistronic expression vector (Clontech) at the NheI/NotI site.
cDNA for full-length ZAP-70 was amplified from the human leukocyte Quick Clone cDNA (Clontech, Palo Alto, CA). The ZAP-70 gene was cloned into a pAcGFP1-N1 vector (Clontech) to fuse an AcGFP1 at its C-terminus. The ZAP-70/AcGFP1 gene was amplified to append EcoRI and PstI sites at their termini, and the PCR product was cloned into pBI-CMV4 at the above sites, exchanging the coding region for DsRed2. The resulting plasmid, termed pBI-ZAP-AcGFP, was digested with NheI and NotI, then the CAR45-28ζ gene was ligated into it (designated pBI-ZAP-AcGFP/CAR45-28ζ).
Expression of CARs in T-cells. Jurkat cells were cultured in an RPMI-1640 medium supplemented with 10% fetal calf serum (FCS), 100 U/ml penicillin and 100 μg/ml streptomycin. The pBI vector containing CAR gene was transfected into the Jurkat cells with the GenePORTER 3000 Transfection Reagent (Genlantis, San Diego, CA).

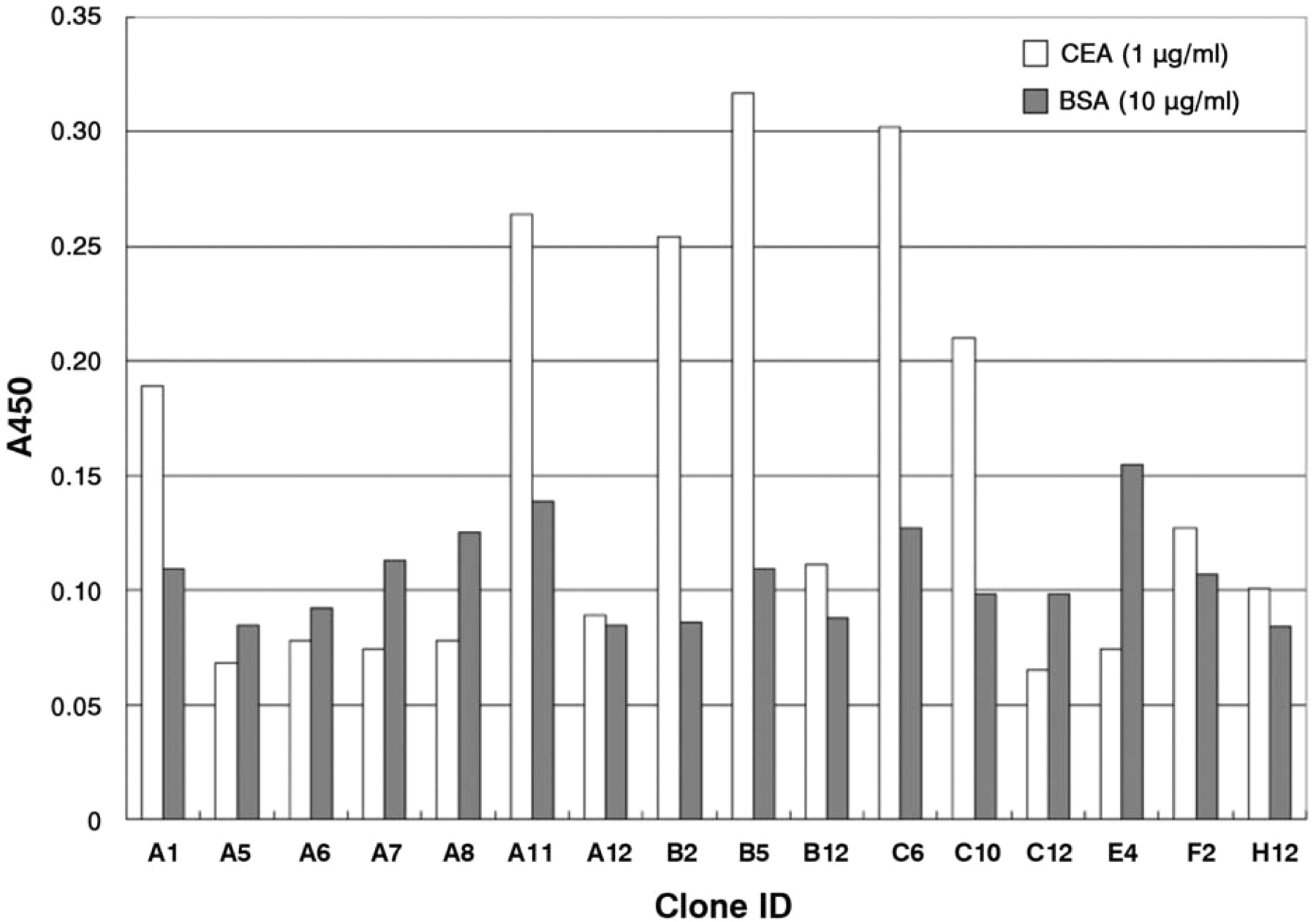
The screening of phage clones displaying anti-CEA scFv by monoclonal phage ELISA. The binding of phages displaying anti-CEA scFv to immobilized CEA (white bar) or BSA (gray bar) was detected by HRP-labeled anti-M13 antibody.
IL-2 secretion assay. Microtiter plates were coated with anti-CD3ε (UCHT1; 500 ng/ml; Beckman Coulter, Miami, FL), anti-CD28 antibody (CD28.2; 500 ng/ml; Beckman Coulter) or CEA (20 μg/ml). Jurkat cells (1×106 cells/ml) were incubated for 24 h at 37°C in coated plates. The culture supernatants were analyzed for secretion of IL-2 using the Human IL-2 ELISA Ready-SET-Go! (eBioscience, San Diego, CA) as per the manufacturer's specifications.
Western blot analysis. The cells were stimulated with immobilized anti-CD3ε alone, anti-CD3ε plus anti-CD28 or immobilized CEA for 15 min at 37°C. The cells were suspended in RIPA buffer at a density of 1×107 cells/ml. The cells were incubated for 1 h on ice, and then boiled in SDS-PAGE sample buffer with 0.1 M dithiothreitol. The samples were separated on 5-20% acrylamide gradient gels and transferred to polyvinylidene fluoride membrane. The blots were blocked with StartingBlock T20 blocking buffer (Thermo Scientific, Bellefonte, PA, USA). The membranes were incubated for 1 h with specific antibody, washed five times in PBS containing 5% StartingBlock, and detected using horseradish peroxidase (HRP)-conjugated secondary antibody. Immunoreactive bands were visualized using the SuperSignal West Pico kit (Thermo Scientific) and then were developed using the LAS3000 chemiluminescence system (Fujifilm, Tokyo, Japan).
Confocal microscopy-assisted cell spreading assay. A cell spreading assay was performed as described previously (15, 16). Eight-chambered coverslips (Lab-Tek II; Nunc) were treated with a 0.01% poly-L-lysine solution (Sigma, St Louis, MO, USA). The slides were coated overnight at 4°C with anti-CD3ε (10 μg/ml). Cells (5×105) were seeded onto the bottoms of the chambers containing RPMI-1640 medium with 10% FCS and 20 mM N-2-hydroxyethyl-piperazine-N'-2-ethanesulfonic acid for 3 min and fixed for 30 min at 37°C by in 8% paraformaldehyde in PBS. The chambers were washed with PBS containing 10 mM glycine, permeabilized for 5 min with 0.1% Triton X-100, rinsed in PBS containing 5% StartingBlock, and blocked for 15 min in 100% StartingBlock. The chambers were incubated for 1 h at room temperature and then overnight at 4°C with anti-CD3ζ (Santa Cruz Biotechnologies, Santa Cruz, CA, USA) and anti-myc antibody (MBL, Nagoya, Japan) pre-labeled with Zenon Alexa Fluor 647 (for goat IgG) or 555 (for rabbit IgG) (Invitrogen, Carlsbad, CA, USA), respectively. The chambers were washed with 5% StartingBlock and re-fixed for 15 min with 4% paraformaldehyde, and washed PBS containing 10 mM glycine. In some experiments, Jurkat cells were preincubated at 37°C for 20 min with the Src kinase inhibitor PP2 (10 μM) (Enzo Life Sciences, Farmingdale, NY). Fluorescent images were acquired on a LSM710 confocal system (Carl Zeiss, Göttingen, Germany).

Nucoleotide and amino acid sequences of anti-CEA scFv from C2-45 hybridoma. Sequences were analyzed using the IMGT/V-QUEST and IMGT/JunctionAnalysis online tools (17), and each complementarity determining region (CDR) of the cloned scFv gene was determined according to Chothia's definition (18). The (Gly4Ser)3 linker sequence is indicated in italics.
Results
Cloning of anti-CEA scFv gene by phage display. The functional screening of VH/VL genes is an essential step for cloning the V-gene encoding a monoclonal antibody since hybridoma cells often express more than one of the VH/VL genes each due to the fusion partner. A phage display technique was employed to directly clone the gene encoding anti-CEA scFv based on its binding activity. The cDNAs of VH and VL were amplified using reverse-transcribed mRNA from C2-45 hybridoma with mixture of leader and constant region-specific primers. The amplified V-gene fragments were assembled into scFv form with (Gly4Ser)3 linker by splice overlap extension PCR to display scFv protein on phage. The scFv genes were ligated into a pIT2 phagemid vector, and used to transform E. coli TG-1 for preparation of scFv-displaying phages. Specific binders were selected by phage ELISA after one round of phage panning on immobilized CEA. Figure 2 shows that some clones showed specific binding against CEA, but negligible binding to BSA, thus indicating that anti-CEA scFv harboring phages were successfully enriched. The nucleotide sequences of positive scFv clones were determined, and it was found that they had essentially the same sequence (Figure 3).

An SDS-PAGE analysis of CAR protein expression. Lysates were prepared from wild-type and CAR-expressing Jurkat cells, and immunoreactive bands were detected by Western blotting using an anti-CD3ζ, as described in the Materials and Methods.
Construction and expression of CAR. Two types of CAR were genetically constructed using cloned anti-CEA scFv gene, as described in the Materials and Methods. One CAR gene, namely CAR45-ζ, contained the CD8 leader sequence, anti-CEA scFv, CD8 hinge region, and transmembrane (TM) and cytoplasmic domains of CD3ζ. Another, CAR45-28ζ, contained the transmembrane and cytoplasmic domains of CD28 instead of CD3ζ TM. Both CAR genes were subcloned into the pBI-CMV4 vector, and the constructs were transfected into Jurkat cells. The protein expression of each CAR was confirmed by Western blotting using an anti-CD3ζ antibody (Figure 4).
CEA-dependent activation of signaling molecules in CAR-transfectants. The activation status of signaling molecules downstream of the TCR was analyzed by immunoblotting with phosphospecific antibodies to investigate the early steps of T cell signal transduction by CAR, Figure 5 demonstrates that CEA stimulation induced protein phosphorylation of ERK, JNK, and p38 in CAR-expressing Jurkat cells. These observations demonstrate that CEA was indeed able to induce the T-cell activating cascades via CAR proteins.
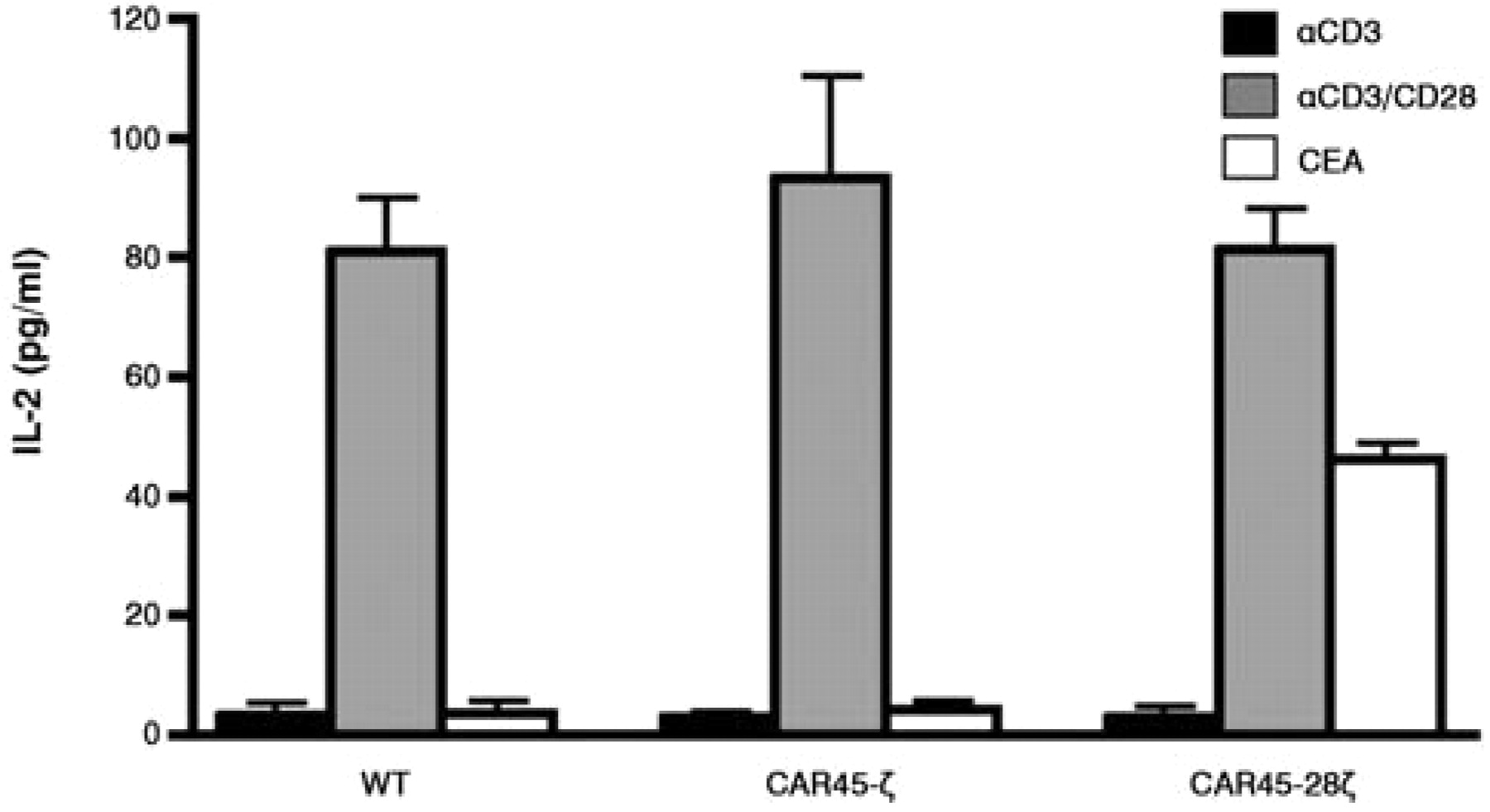
IL-2 production of CAR-directed Jurkat cells. CAR-mediated T-cell activation was investigated by an IL-2 secretion assay to assess whether the CARs constructed were functional. No IL-2 secretion was observed in CAR45-ζ-expressing cells when CEA was used as a stimulant, nor in wild-type cells stimulated with anti-CD3 alone. In contrast, CEA was clearly able to induce the production of IL-2 in CAR45-28ζ-transfectants (Figure 6), thus indicating that the CD28 cytoplasmic domain in CAR45-28ζ was able to functionally deliver the co-stimulating signal and activate the T-cell signaling pathway(s) required for the production of IL-2 by CEA. This CAR45-28ζ was then used in the subsequent imaging study.
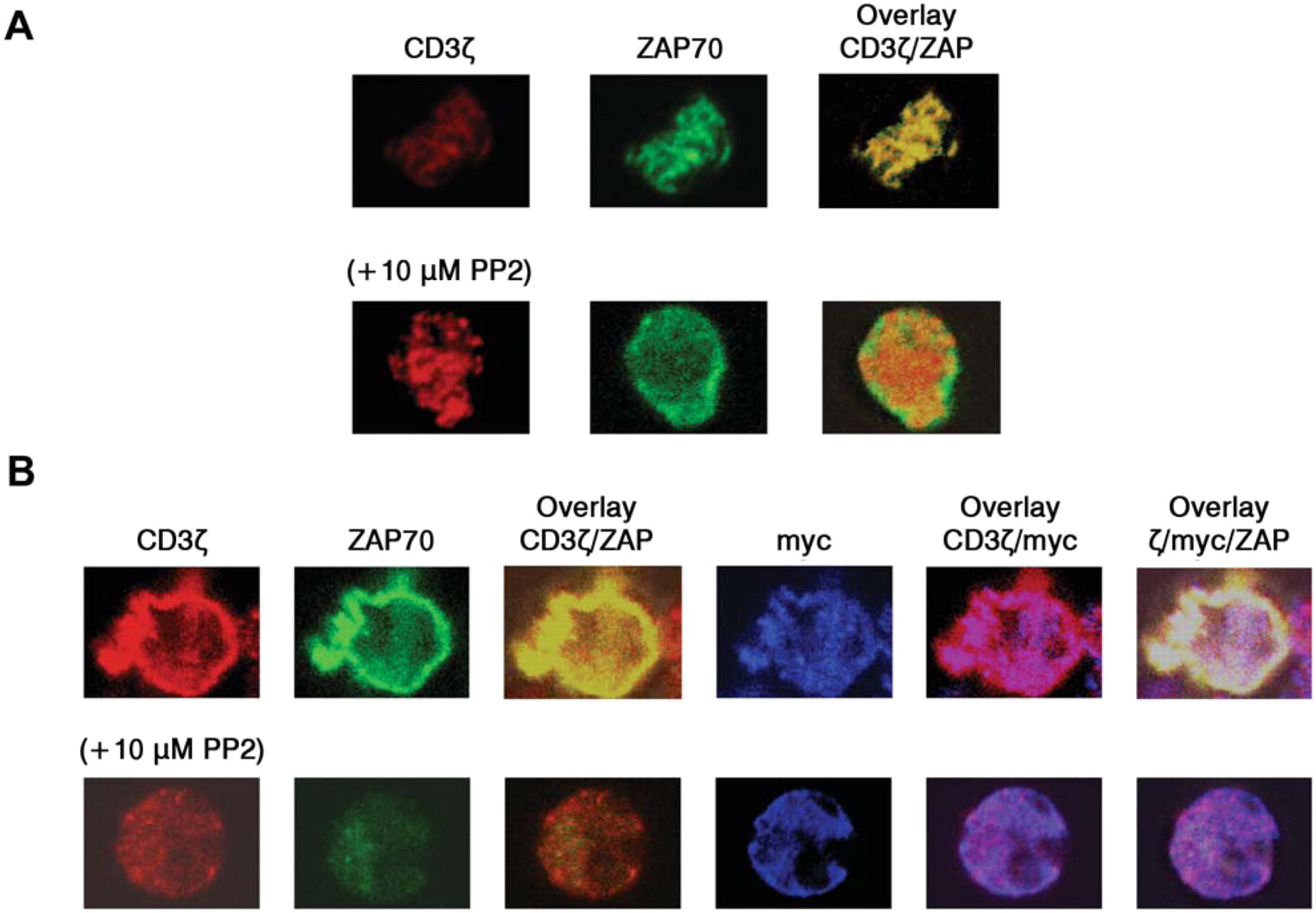
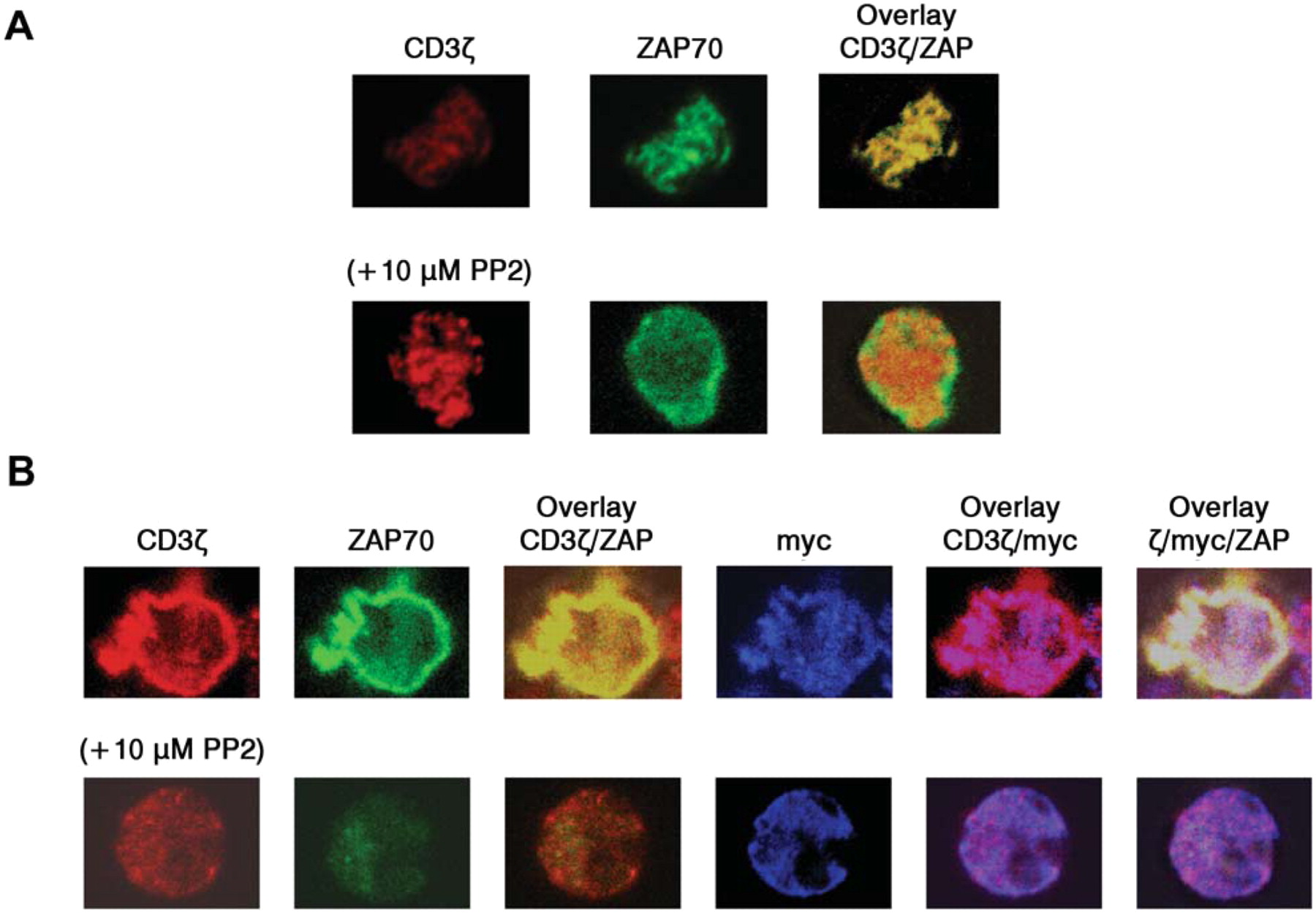
CEA-dependent formation of CAR-containing signaling clusters. A T-cell spreading assay was carried out, utilizing the AcGFP-tagged ZAP-70 to visualize a formation of immunoreceptor tyrosine-based activation motif (ITAM)-mediated signaling clusters. Cells were dropped onto a surface coated with a stimulatory antibody or CEA. This might result in receptor clustering and recruitment of signaling molecules to the points of contact with the stimulatory surface (15, 16). Endogenous TCRs detected with anti-CD3ζ were observed in ZAP-containing clusters when Jurkat cells expressing ZAP-AcGFP alone were introduced onto anti-CD3ε-coated coverslips (Figure 7A, upper panels). Treatment with PP2, an inhibitor of Lck and Fyn, prevented the recruitment of ZAP-70 into the clusters without inhibiting the clustering of the TCR, as previously reported (Figure 7A, lower panels). As expected, no cells attached to the coverslips when CEA was used as a coating protein (data not shown). However, myc-tagged CAR45-28ζ-expressing cells did adhere to the CEA-coated surface, and the clear co-localization of CAR45-28ζ and ZAP-70 was observed (Figure 7B). Note that immunofluorescent signals with anti-CD3ζ antibody show the localization of endogenous TCRs as well as that of CAR45-28ζ receptors. These results indicate that CAR45-28ζ was able to induce the formation of TCR-like signaling clusters with its corresponding antigen CEA.
Discussion
The human anti-CEA scFv gene was cloned using the phage display technique and fully human CARs, CAR45-ζ and CAR45-28ζ were constructed. These CARs were successfully expressed in Jurkat cells and were activated with CEA stimulation. Co-stimulation signal via the cytoplasmic domain of CD28 was requisite for efficient IL-2 production in CAR-expressing T-cells (Figure 6), consistent with previous reports (8, 19). However, the phosphorylation profiles of the signaling proteins examined after CEA stimulation were not obviously different between CAR45-ζ and CAR45-28ζ (Figure 5). The immunofluorescent study showed that CAR45-28ζ induced the formation of signaling clusters at the CAR/CEA interaction interface (Figure 7). Of course, the anti-CEA scFv recognizes a unique epitope, and the binding of CAR to CEA is thought to be monovalent. On the other hand, the stimulating monoclonal anti-CD3 antibody has two antigen-binding sites. Such a difference of the avidity might affect the activity of receptors for molecular clustering. A previous report showed that that soluble CEA was not able to effectively inhibit the antitumor activity of a chimeric antibody containing anti-CEA scFv (20), thus it is likely that the use of immobilized CEA as a stimulant helped to assemble CAR45-28ζ and ZAP-70.

The phosphorylation status of the signaling proteins in CAR-expressing Jurkat cells. Cells were activated for 15 min with the indicated stimulant, and the lysates were used for the Western blot analysis. Untransfected cells were used as a negative control. Active forms of ERK, JNK, and p38 were detected using phospho-specific antibodies (Anti-ACTIVE®; Promega).
The present study demonstrated that a properly designed chimeric T-cell receptor can trigger a wild-type TCR-like signaling event at the molecular level. It would therefore be interesting to see how long the CAR-containing clusters are stable and where the clusters move in comparison to those formed by wild-type TCR because the lifetime and dynamics of the signaling clusters are crucial for the activation and fate of T-cells (21, 22). Such questions could be answered by real-time live cell imaging of CAR-grafted cells. The current molecular imaging approach to CAR would provide useful clues for further improvement and optimal design of CAR constructions.
It is necessary to determine whether CEA-positive tumor cells are actually killed by the primary T-cells bearing the CAR constructed in the current study. A viral transfer technique, such as retro- or lentivirus-based vectoring system, would be needed to achieve a sufficient efficiency of gene transfer into quiescent T-cells. Such experiments are now currently underway.
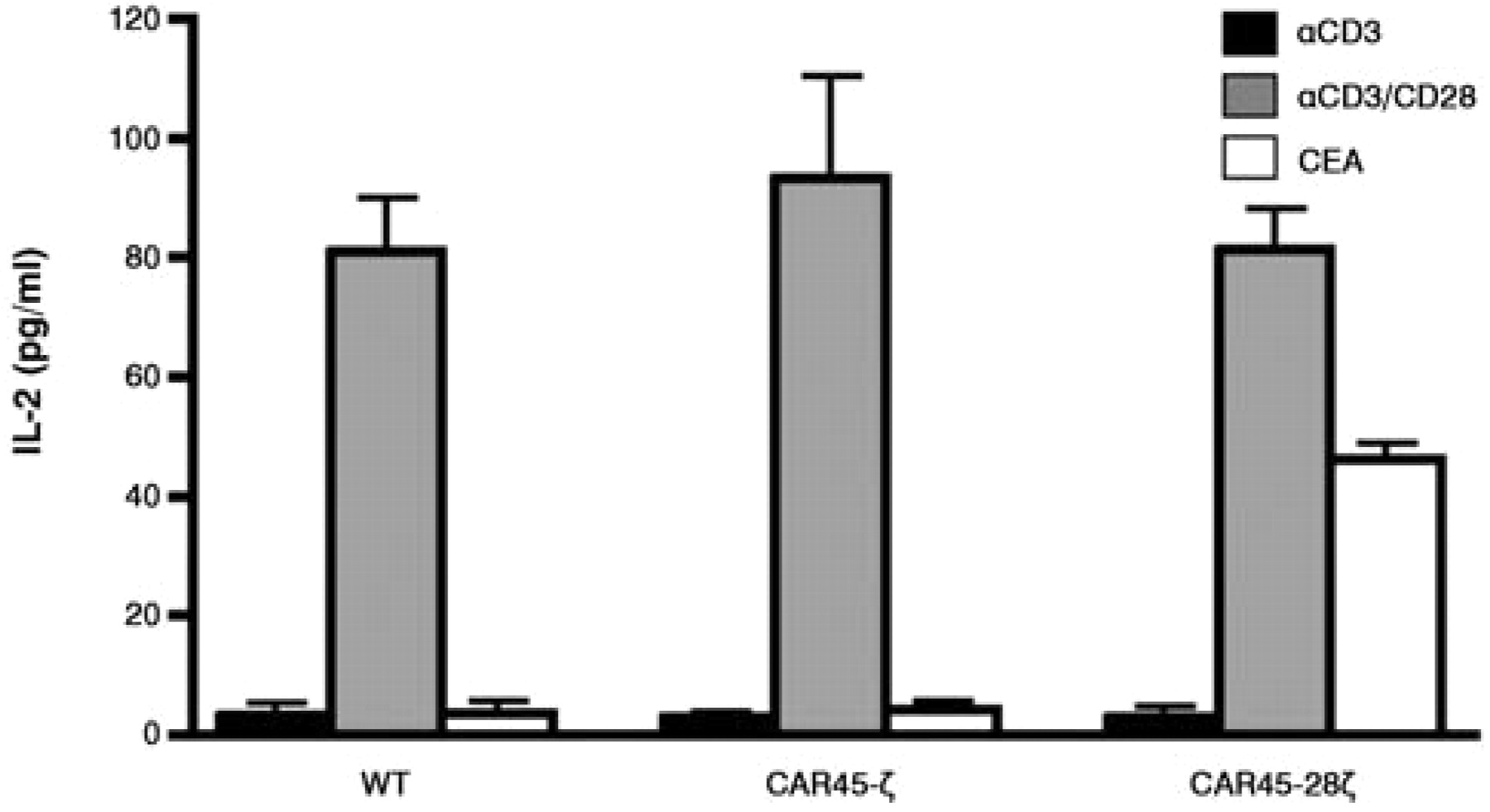
IL-2 secretion from CAR-expressing Jurkat cells. The concentration of IL-2 in the cell supernatant was measured 24 h after stimulation with anti-CD3, anti-CD3 plus anti-CD28, or CEA.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The formation of the signaling clusters in CAR-expressing cells by antigen stimulation. A: Jurkat cells expressing ZAP-70-AcGFP were plated on stimulatory coverslips coated with anti-CD3ε, then fixed and stained with fluorescently labeled anti-CD3ζ. Confocal images at the interface between the attached cell and the stimulatory surface were collected. Lower panels show images from PP2-treated cells. B: Jurkat cells co-expressing ZAP-70-AcGFP and CAR45-28ζ were plated onto a CEA-coated coverslips, fixed and stained with Alexa-labelled anti-CD3ζ and anti-myc antibodies.
Acknowledgements
This work was supported by a Grant-in-Aid for Scientific Research (C) from the Japan Society for the Promotion of the Science.
- Received March 25, 2010.
- Revision received June 10, 2010.
- Accepted June 16, 2010.
- Copyright© 2010 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved