

Abstract
Background: It is suggested that the phosphatidylinositol 3-kinase (PI3K/Akt) pathway may lead to tamoxifen (Tam) resistance in the estrogen receptor-α (ER-α)-positive breast cancer cell line, MCF-7. Our previous results demonstrated that keratinocyte growth factor (KGF) down-regulates ER-α expression and increases Tam resistance in MCF-7 cells. Therefore, we hypothesized that a possible mechanism for developing Tam resistance could be the regulation of ER-α and Bcl-2 family proteins through modulation of Akt activity. Materials and Methods: MCF-7 cells were treated with KGF, LY294002 (LY), a PI3 kinase inhibitor, 4-OH-Tam, KGF with LY, KGF with LY and 4-OH-Tam, or vehicles as control for 24 hours. Total RNA was extracted from MCF-7 cells and real-time PCR was employed to identify the ER-α expression in response to KGF. To determine that the resistance to 4-OH-Tam-inducing cell killing after the KGF treatment was due to the inactivation of the apoptotic pathway, low molecular weight DNA was isolated from cells of different treatments and inter-nucleosomal DNA fragmentation was investigated. The phosphorylation of signaling intermediates Akt, Bad, the activation of caspase-9, and the expression of ER-α, Bcl-2, Bcl-xL, and Bax were evaluated by immunoblot analysis for the study of KGF signaling effects. To determine the involvement of PI3K/Akt pathway in the survival effect of KGF, the growth rate of MCF-7 cell was measured by non-radioactive cell proliferation assay after treatments of KGF, LY, 4-OH-Tam, KGF with LY, KGF with LY and 4-OH-Tam, or vehicles as control for 3 days. The results of real-time PCR and cell proliferation assay were analyzed by Student's t-test and p-values of less than 0.05 were considered statistically significant. Results: Our results showed that in MCF-7 cells KGF increased Akt phosphorylation and induced ER-α mRNA expression which could be blocked by a PI3K/Akt pathway inhibitor, LY. KGF treatment also induced apoptosis based on the observation of the suppression of DNA fragmentation, variable increase in the expression of the Bcl-2 and Bcl-xL proteins and the decrease of the active form of caspase-9 protein, whereas LY blocked the anti-apoptotic effects of KGF. In the cell proliferation assay, KGF maintained MCF-7 cell survival in the presence of 4-OH-Tam which could be blocked by LY. Conclusion: We confirmed the regulation of ER-α by KGF in human breast cancer cells at both mRNA and protein levels. We further demonstrated that KGF may play an inhibitory role in the induction of breast cancer cell apoptosis, conferring resistance against anticancer drugs on breast cancer cells.
- KGF
- estrogen
- tamoxifen
- ER-α
- PI3/Akt
- breast cancer
- apoptosis
- Bcl-2 family
Established evidence derived from in vitro, in vivo and epidemiological studies have strongly implicated estrogen as a carcinogen for the development and progression of breast cancer (1, 2). The tumorigenic effects of estrogens are mediated primarily through interaction with the estrogen receptor-α (ER-α). Estrogen binding to ER-α activates the protein through phosphorylation, dissociates chaperon proteins such as heat-shock protein 90 and alters its conformation (3). Hormone-bound ER-α then dimerizes and binds to estrogen response elements (EREs) within promoters of estrogen-regulated genes directly or indirectly. Many of the estrogen-regulated genes are important for cell proliferation, inhibition of apoptosis, stimulation of invasion and metastasis, and promotion of angiogenesis (4).
Motivated by the mechanisms of action of estrogen, antiestrogenic agents are designed to work by their interaction with the ER-α for the management of estrogen-responsive human breast cancer. Tamoxifen (Tam), a selective estrogen receptor modulator (SERM) has been widely used in the treatment of breast cancer patients (5) and for reducing breast cancer incidence in high-risk populations, (6, 7). Tam has been attributed to cause both growth arrest and apoptosis in breast cancer cells (8). It is believed that the growth arrest effect of Tam is via down-regulation of estrogen-regulated growth factors such as epidermal growth factor (EGF) and transforming growth factor (TGF) in ER-α-positive human breast cancer cells. The apoptotic effect of Tam has been implicated to be mediated by several mechanisms. Tam has been demonstrated to stimulate the activity of caspases 9 and 3 in ER-α-negative human breast cancer cells suggesting the caspase cascade as a potential regulatory mechanism (9, 10).
In spite of the widespread use of Tam and its significant contribution to the reduction in mortality from human breast cancer over the last decade (11), acquired drug resistance, primarily Tam resistance, exists in human breast cancer patients. Accumulating evidence suggest an intricate network of cross-talk between the ER-α and growth factor signalling as a mechanism leading to Tam resistance. The network may consist of peptide growth factors, their receptor tyrosine kinase (12, 13) and downstream signaling proteins such as c-Src, Erk1/2, MAPK (14), protein phosphatase and Akt. The cross-talk between the ER-α and growth factor receptors occurs in several directions. First, ER-α can generate multiple growth-promoting signals via estrogen-induced genes encoding growth factors, their receptors, and other signaling molecules which can provide cell proliferations and survival stimuli (15, 16). Second, phosphorylation of ER-α in the N-terminal region by signal proteins such as MAPK and Akt alters estrogen-regulated gene transcription. Finally, membrane ER-α can activate growth factor receptor tyrosine kinases, such as members of the EGF receptor family (17).
Keratinocyte growth factor/fibroblast growth factor-7 (KGF/FGF-7) is a member of FGF family, which comprises at least 22 members sharing high affinity for heparin as well as high sequence homology with the central core domain of 120 amino acids, which interact with fibroblast growth factor receptors (FGFRs). KGF has a stromal origin and appears to act specifically on epithelial cells in human tissue and is therefore an exclusively paracrine growth factor in human tissues (18, 19). Reports have documented the importance of KGF in controlling mammary epithelium. KGF stimulate normal human breast and human breast cancer epithelial cell proliferation in a dose-dependent manner (20). In rodents, KGF induced the growth of murine mammary epithelial cells in a collagen gel matrix in a heparin-independent manner (21), and systemic administration of KGF to rodents resulted in mammary epithelial hyperplasia (22, 23). It is also suggested that KGF stimulates the migration of breast cancer cells (24-26).
Four functional genes have been identified within the FGFR family including FGFR-1, FGFR-2, FGFR-3, FGFR-4 (27). FGFR-2IIIb, also known as KGF receptor (KGFR) is a glycoprotein with two or three immunoglobulin (I)-like domains, a transmembrane region and a tyrosine kinase catalytic site in cytoplasm. FGFR-2IIIb, which is expressed in epithelial cells, binds specifically to KGF (28, 29). The signal transduction of KGF/KGFR can proceed via Ras/MAPK, and PI3K/Akt pathways (25, 30-34). The findings on KGF stimulation of cell proliferation and motility via Ras/MAPK suggest that KGF/KGFR could potentially influence the development and progression of breast cancer (24, 25). However, the PI3K/Akt pathway by which KGF and KGFR are involved in human breast cancer is not yet fully understood at the present time.
Akt, also known as protein kinase B (PKB), is a serine/threonine kinase (which can be activated by a variety of stimuli including hormones and growth factors). Three Akt members have been identified in humans which include AKT1, AKT2 and AKT3 (35). All three Akt members contain a pleckstrin homology (PH) domain in their amino-terminal region, a central kinase domain, and a regulatory domain in the carboxy-terminal region (36). Akt activation occurs in a phosphoinositide-3 kinase (PI3K) dependent manner. PI3K-activated PIP3 acts as lipid second messengers for the translocation of Akt and phosphoinositide-dependent kinase (PDK) to the plasma membrane, where PDK mediates Akt phosphorylation on threonine T308 and serine S473 (37, 38). Recent studies have shown that activation of Akt protein is able to regulate cell survival through phosphorylation and expression of Bcl-2 family proteins (35). In the apoptotic pathway, members of the Bcl-2 family are mitochondrial integrity regulators followed by cytochrome c release, activation of the apoptotic protease-activating factor (Apaf-1) and the cysteine protease, caspase-9 to initiate a caspase cascade (9).
Hormones and growth factors such as estradiol-17β (E2), herregulin β1 (HRGβ1), EGF and insulin-like growth factor-I (IGF-I) have been demonstrated to activate the PI3K/Akt pathway and regulate ER-α expression in the hormone-dependent human breast cancer cell line, MCF-7 (13, 39, 40). Our laboratory also has been able to show that KGF can regulate ER-α expression and Tam resistance in MCF-7 cells (41). The goal of the present study was to identify the mechanisms for the KGF-induced Tam resistance displayed by ER-α positive tumors. Our hypothesis is that Tam resistance induced by KGF are mediated by the ER-α expression and apoptosis through the activation of the PI3K/Akt pathway.
Materials and Methods
Cell culture. MCF-7, a human breast cancer cell line, was purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). The cells were maintained in Dulbecco's modified Eagle's medium and Ham's F12 medium (1:1) (DMEM/F12) mixture, containing no phenol red (Sigma Chemical Co., St Louis, MO, USA), supplemented with 5% fetal bovine serum (FBS, GIBCO Cell Culture™, Carlsbad, CA, USA) and antibiotic-antimycotic (100 U/ml penicillin G sodium, 100 μg/ml streptomycin sulfate and 0.25 μg/ml amphotericin B, GIBCO Cell Culture™). MCF-7 cells were seeded and cultured in 75-cm2 culture flasks in a humidified incubator (5% CO2:95% air, 37°C). The media were replaced every 2 days. At 85% confluence, cells were washed twice with calcium- and magnesium-free phosphate-buffered saline (PBS, pH 7.3). Cells were then trypsinized with 1% trypsin-5.3 mM EDTA (GIBCO Cell Culture™) in PBS for 10 min at 37°C. The trypsinization was stopped by addition of culture medium containing 5% FBS. Cells were then centrifuged and re-suspended in the original cultured medium as described above and subcultured into 75-cm2 culture flasks at a ratio of 1:3.
Total RNA extraction and reverse transcription-polymerase chain reaction (RT-PCR). Recombinant KGF was purchased from PeproTech Inc. (Rocky Hill, NJ, USA). LY294002 (LY) was purchased from Cell Signaling Technology Inc. (Beverly, MA, USA) and, 4-hydroxy Tam (4-OH-Tam) was purchased from Sigma Aldrich Inc. (Milwaukee, WI, USA). Before treatment, MCF-7 cells were seeded in DMEM/F12 supplemented with 5% FBS in 24-well culture plates at a density of 2×104 overnight. The media were changed to DMEM/F12 supplemented with 0.02% bovine albumin Fraction V (GIBCO Cell Culture™) for 24 hours. MCF-7 cells were treated with 20 ng/ml KGF, 10 μM LY, 20 ng/ml KGF with 10 μM LY and 20 ng/ml KGF, or vehicles as control in DMEM/F12 supplemented with 5% dextran-coated charcoal (DCC) (Dextran T-70, Pharmacia Biotech, Uppsala, Sweden; activated charcoal, Sigma Aldrich Inc.)-treated FBS for 24 hours. Total RNA was extracted from MCF-7 cells by using the Trizol® Reagent (Invitrogen™, Carlsbad, CA, USA). RT-PCR was performed in a gradient mastercycler (Eppendorf, Scientific Inc., Westbur, NY, USA). For cDNA synthesis, 1 mg of total RNA from cells was reverse-transcribed in a final volume of 50 ml containing 200 U M-MLV Reverse Transcriptase (Invitrogen™) 10 ml 5× 1st strand buffer (15 mM MgCl2, 375 mM KCl, 250 mM Tris-HCl pH 8.3), 0.2 mM of dATP, dCTP, dGTP and dTTP, 0.01 M DTT, 1 U of RNA Guard RNase inhibitor (Pharmacia Biotech) and 1 mM random hexamers. Samples were incubated at 65°C for 5 min and 37°C for 50 min, and reverse transcriptase was inactivated by heating at 70°C for 15 minutes.
Relative quantification of ER-α expression and real-time PCR. The nucleotide sequences of the hybridization probes and primers for the ER-α and 36B4 genes were as follows: ER-α genes: sense AGCTCCTCCTCATCCTCTCC, anti-sense TCTCCAGCAGCAGG TCATAG and probe FAM-TCAGGCACATGAGTAACAAAGGCA-TAMRA from TaqMan® (Foster, CA, USA); 36B4 gene: sense CTGGAGACAAAGTGGGAGCC, anti-sense TCGAACACCTGC TGGATGAC and probe FAM-ACGCTGCTGAACATGCTCAAC ATCTCC-TAMRA from Synthegen (Houston, TX, USA).
The comparative CT method was used for the relative quantitation of ER-α expression. The parameter CT (threshold cycle) is defined as the fractional cycle number at which the fluorescence generated by cleavage of the TaqMan® probe-amplicon complex formation passes a fixed threshold above baseline. The relative ER-α gene expression level was expressed as 2-(ΔCT sample-ΔCT control) = 2-(ΔΔCT). The ΔCT was a normalized value of ER-α CT value to the endogenous control of 36B4 CT value. All PCR reactions were performed using an ABI Prism 7000 Sequence Detection System (Perkin-Elmer Applied Biosystems, Boston, MA, USA). For each PCR run, a master-mix was prepared on ice with 10 × Real-Time PCR Buffer (Ambion®, Austin, TX, USA), 25 mM MgCl2, 10 mM dATP, dCTP and dGTP, and dUTP, 5 mM of each primer, 100 mM TaqMan® probe and 5 U/μl of Platinum® Taq DNA polymerase (Invitrogen™). Five micro liters of each cDNA sample were added to 50 ml of the PCR master-mix. The thermal cycling conditions comprised an initial step at 50°C for 2 minutes, 95°C for 10 minutes, and 45 cycles at 95°C for 15 seconds followed by annealing at 60°C for 1 minute.
Low molecular weight DNA isolation and gel electrophoresis. To study the appearance of DNA ladders, low molecular weight DNA was isolated. This method enables the detection of DNA ladders with high sensitivity. MCF-7 cells were seeded in 6-well culture plates with DMEM/F12 supplemented with 5% FBS at a density of 2×105 overnight. The media were changed to DMEM/F12 supplemented with 0.02% bovine albumin Fraction V (BSA) (GIBCO®, Grand Island, NY, USA) for 24 hours. MCF-7 cells were then treated with 20 ng/ml KGF, 10 μM LY, 2.5 to 10 μM 4-OH-Tam, 20 ng/ml KGF with 10 μM LY, 20 ng/ml KGF with 10 μM LY and 10 μM 4-OH-Tam, or vehicles as control in DMEM/F12 supplemented with 0.02% BSA for 48 hours. All cells including floating cells were fixed in cold 70% ethanol for overnight. After removal of ethanol, the cell pellet was re-suspended in 40 ml PC buffer (mixture of 192 parts of 0.2 M Na2HPO4 and 8 parts of 0.1 M citric acid, pH 7.8) and left at room temperature for 30 minutes. The pellet was spun down at 10,000 rpm for 5 minutes and the supernatant was transferred into a fresh tube. Subsequently, 3 ml of NP-40 (0.25% in distilled water) and 3 ml of RNase A (1 mg/ml in distilled water) were added and this was incubated at 37°C for 30 minutes. After adding 3 ml of Proteinase K solution (1 mg/ml), the mixture was incubated for a further 30 minutes at 37°C; 15 ml DNA solution were then separated on a 0.8% agarose gel at 100 V for 1.5 hours.
Western blot analysis. MCF-7 cells were seeded in 6-well culture plates with DMEM/F12 supplemented with 5% FBS at a density of 2×105 overnight. The media were changed to DMEM/F12 supplemented with 0.02% bovine albumin Fraction V (BSA) (GIBCO®, Grand Island, NY, USA) for 24 hours. For the evaluation of KGF-induced protein phosphorylation, MCF-7 cells were pre-treated with 10 μM LY or vehicles as control in DMEM/F12 supplemented with 0.02% BSA for 30 minutes. MCF-7 cells were then treated with 1 to 20 ng/ml KGF, 10 μM LY, 20 ng/ml KGF with 10 μM LY, or vehicles as control in DMEM/F12 supplemented with 0.02% BSA for a time from 0 to 30 minutes. For the evaluation of KGF-induced protein expressions, MCF-7 cells were treated with 1 to 20 ng/ml KGF, 10 μM LY, 10 μM 4-OH-Tam, 20 ng/ml KGF with 10 μM LY, 20 ng/ml KGF with 10 μM LY and 10 μM 4-OH-Tam, or vehicles as control in DMEM/F12 supplemented with 0.02% BSA for 24 hours. Total proteins were then extracted from MCF-7 cells with MPER® (Pierce Biotechnology, Rockford, IL, USA) according to manufacturer's instructions. The lysates were heated to 95-100°C for five minutes, loaded on a 4-15% Tris-HCl Ready Gel (BIO-RAD Laboratories, Hercules, CA, USA) in equal amounts of protein for electrophoresis. Proteins were transferred onto the polyvinylidene fluoride (PVDF) membranes (Millipore, Bedford USA). The membranes were blocked by immersing in 5% non-fat milk in PBS containing 0.1% Tween 20 (PBS-T) at 4°C overnight. The membranes were then incubated in PBS-T and incubated for one hour with the primary antibody. Primary antibodies used for immunoblotting were to ER-α (HC-20) actin (C-11), Bcl-2 (Santa Cruz Biotech, Heidelberg, Germany), Akt, phospho-Akt (p-Akt), cleaved caspase-9, phospho-Bad (p-Bad), Bax, and Bcl-xL (Cell Signaling Technology, Inc.). After washing in PBS-T, the membranes were incubated with the horseradish peroxidase-linked secondary anti-goat or anti-rabbit immunoglobulin antibody for 1 hour at room temperature followed by PBS-T wash. After washing in PBS-T, the proteins were visualized with a chemiluminescent detection system (ECL; Amersham Pharmacia Biotech, Piscataway, NJ, USA) and exposed to a cooled CCD camera (Fujifilm Medical System U.S.A. Inc., Stamford, CT, USA).
Cell proliferation assay. MCF-7 cells were seeded in 96-well culture plates with DMEM/F12 supplemented with 5% FBS at a density of 1×103 in a volume of 100 μl/well overnight. After cells were attached to the wells, the medium was replaced with 100 μl of DMEM/F12 containing 1% of DCC FBS. Cells were then treated with 10 μM 4-OH-Tam, 20 ng/ml KGF, 10 μM LY, 10 μM 4-OH-Tam plus 20 ng/ml KGF, the combination of 10 μM 4-OH-Tam, 20 ng/ml KGF and 10 μM LY, or the vehicle as control in the same fresh medium for 48 hours. Experiments were performed in 4 replicate wells for each group. The cell proliferation rate was quantified by using CellTiter™ AQueous assay (Promega, Madison, WI, USA). Briefly, at the end of treatment, 100 μl of fresh medium with 20 ml of freshly combined MTS/PMS (the ratio of MTS: PMS is 20:1)) solution was added to each well. The plates then were incubated for 1.5 h. The optical density was read at 490 nm (OD490 nm) by an ELISA plate reader (Molecular Devices, Menlopark, CT, USA).
Statistical analysis. The results for the PCR reaction and for Western blot analysis of the expression of Bcl-xL, Bcl-2, and Bax proteins are presented as the mean±standard deviation (s.d.) for three replicates as one group. The non-radioactive cell proliferation assay is presented as the mean±standard deviation (s.d.) for four replicate culture wells as one group. Analysis was performed by using StatView statistical software for Windows (SAS Institute Inc., Cary, NC, USA). Statistical differences were determined by using Student's t-test for independent groups. P-values of less than 0.05 were considered to be statistically significant.
Results
KGF stimulated Akt phosphorylation. We used total Akt which was not phosphorylated by KGF as an internal control to measure the fold changes of Akt phosphorylation. We found that KGF induced increases of Akt phosphorylation by 2.6- and 2.4-fold at concentrations of 10 and 20 ng/ml, respectively (Figure 1A). The time course of the activation of Akt by 20 ng/ml KGF is given in Figure 1B. KGF induced Akt activation rapidly in 5 minutes and reached a peak at 15 minutes. Concentrations of KGF that stimulated Akt activations did not trigger the Erk 1/ 2 activities (data not shown). Western blot analyses showed that KGF rapidly stimulated phosphorylation of Akt but not that of Erk1/2 in MCF-7 cells.
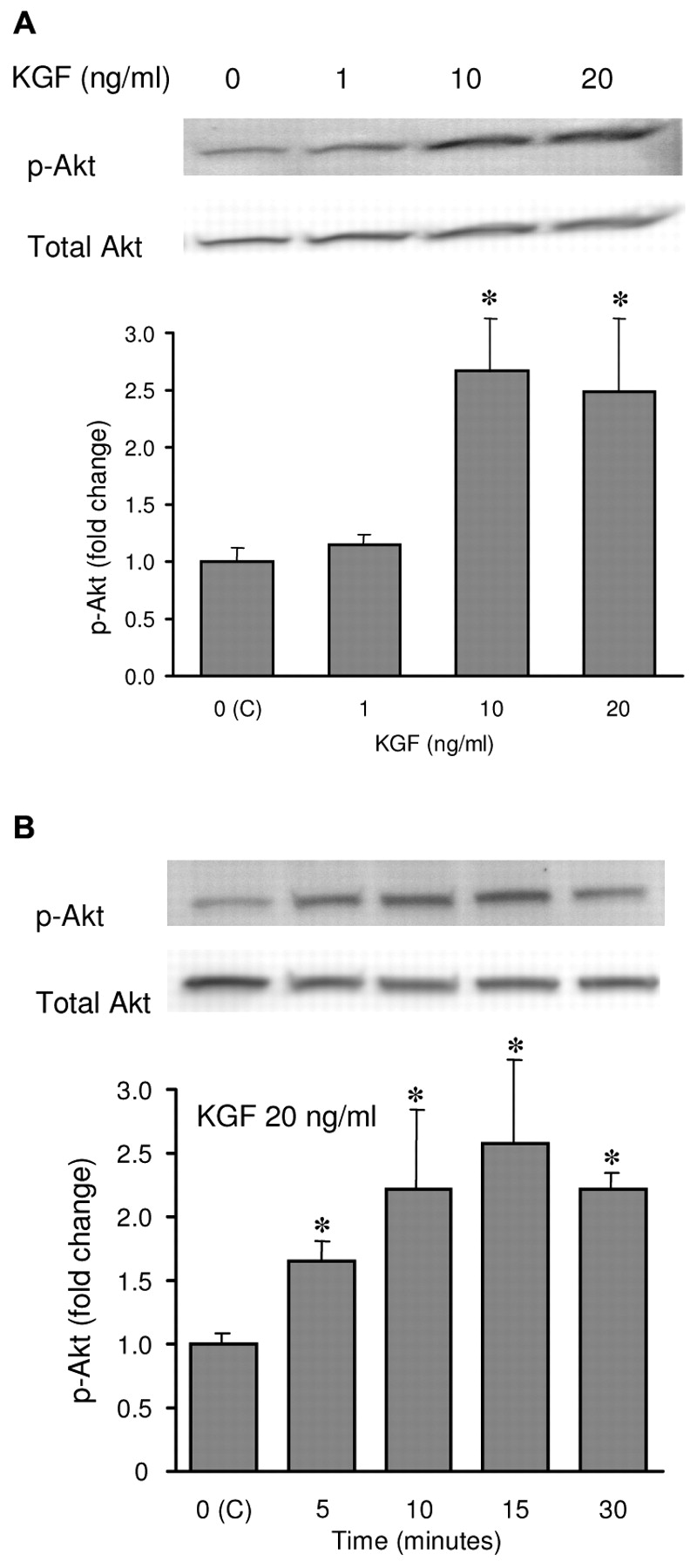
KGF stimulated Akt phosphorylation. A, MCF-7 cells were serum-starved for 24 hours and treated with 1, 10 and 20 ng/ml KGF for 20 minutes. B, MCF-7 cells were serum starved for 24 hours and treated with 20 ng/ml for the times indicated. Western blot of cell extracts were probed using anti-phospho-Akt (p-Akt, Ser 473) or total anti-Akt antibody. Total Akt was used as a control for equal loading and transfer. The fold change between treatments was determined by densitometry and results were present as a percentage of that of the control (C). Results represent the mean value of three independent experiments ± s.d. Asterisks represent the significant difference (p<0.05) from the control group.

LY inhibited KGF-induced Akt phosphorylation in MCF-7 cells. Cells were serum starved for 24 hours and treated with 20 ng/ml KGF in the presence or absence of 10 μM of LY. Western blot of cell extracts are probed using anti-phospho-Akt (p-Akt, Ser 473) or total anti-Akt antibody. Total Akt was used as a control for equal loading and transfer. The ratio between phosphorylated and total Akt was determined by densitometry and results are represented as a percentage of that of control (C). Results represent the mean value of three independent experiments ± s.d. The asterisk represents the significant difference (p<0.05) of the KGF-treated group from the control group.
Akt induction by KGF required PI3K. In order to confirm that phosphorylation of Akt required the activation of p85 and p110 PI3K, we evaluated KGF-induced Akt phosphorylation in the presence of a PI3K inhibitor, LY in MCF-7 cells. Under the presence or absence of the LY (10 μM) pre-treatment for 30 minutes, MCF-7 cells were then treated with 20 ng/ml KGF for 10 minutes. Inhibition of PI3K by LY resulted in a substantial reduction of Akt kinase phosphorylation compared to cells treated with vehicle alone (Figure 2). These data provide the evidence of activation of Akt kinase by KGF via a PI3K-mediated pathway.
PI3K inhibitor blocked KGF-induced ER-α gene expression. Our lab previously reported that KGF acted as a survival factor via modulation of ER-α gene expression in the human breast cancer cell line, MCF-7 (41). Our results here demonstrated that 20 ng/ml KGF reduced ER-α protein and mRNA by 60% and 56%, respectively. LY reversed the effect of KGF on ER-α expression (p<0.05) (Figures 3 and 4). These results demonstrated that KGF regulated ER-α expression through the PI3K/Akt pathway, which could lead to an increase of anti-estrogenic resistance in MCF-7 cells.

PI3K inhibitors blocked KGF-induced ER-α gene expression. MCF-7 cells were seeded in DMEM/F12 without phenol red supplemented with 5% FBS. The media were replaced with DMEM/F12 without phenol red supplemented with 5% DCC FBS and cells were treated with 20 ng/ml KGF in the present or absence of 10 μM LY for 24 hours. At the end of treatment, total RNAs were collected. ER-α mRNA expression was detected by real-time PCR. Results represent the mean value of three independent experiments ± s.d. The asterisk represents the significant difference (p<0.05) from the control group.

PI3K inhibitors blocked KGF-induced ER-α protein expression. MCF-7 cells were seeded in DMEM/F12 without phenol red supplemented with 5% FBS. The media were replaced with DMEM/F12 and without phenol red supplemented with 5% DCC FBS and cells were treated with 20 ng/ml KGF in the present or absence of the 10 μM LY for 24 hours. At the end of treatment, total proteins were collected. ER-α protein was detected by Western blot. The ratio between ER-α and actin was determined by densitometry and results were represented as a percentage of that of control (C). Results are the mean value of three independent experiments ± s.d. The asterisk represents the significant difference (p<0.05) from the control group.
PI3K/Akt dependency of KGF protected MCF-7 cells from apoptosis. Increasing evidence suggests that Tam resistance is associated with growth factors and kinase pathways of these factors (3, 42). Previous results from our laboratory demonstrated that the KGF pathway mediated the effects of Tam on the survival in the MCF-7 cells in a cell proliferation assay (41). These results implied that KGF may protect cells from 4-OH-Tam-induced cell killing. To further determine that the resistance of 4-OH-Tam-inducing cell killing after KGF treatment was due to the inactivation of the apoptotic pathway, we investigated the inter-nucleosomal DNA fragmentation, one of the characteristics of cell apoptosis. The appearance of DNA ladders was observed in cells treated with 10, but not 2.5 or 5 μM of 4-OH-Tam at 48 hours, while untreated cells did not exhibit DNA ladders (Figure 5A). We also found that a significant decrease in DNA ladder formation when cells were treated with the combination of 20 ng/ml KGF and 10 μM 4-OH-Tam. We next investigated the mechanisms involved in the anti-apoptotic effect of KGF. Studies have shown that KGF induced Akt kinase activity and inhibited apoptosis in A549 lung epithelial cells (30). Therefore, we would like to address the question of whether Akt kinase is also involved in KGF-induced anti-apoptotic effects in MCF-7 cells. We incubated the cells in the presence or absence of LY pre-treatment at a concentration of 10 μM. Our results showed that LY abolished the protective effect of KGF in cells exposed to 4-OH-Tam (Figure 5B). These results suggested that KGF contributed to the acquired Tam resistance in human breast cancer cells and the KGF-induced resistance was mediated via the PI3K/Akt pathway.
Pro- and antiapoptotic targets of KGF/PI3K/Akt in MCF-7 cells. To investigate whether the KGF-induced Akt pathway affects pro- and/or anti-apoptotic proteins in the human breast cancer cell line, MCF-7 were treated with serial doses of KGF and various members of the Bcl-2 family were subjected to western blot analysis including anti-apoptotic effectors such as Bcl-2 and Bcl-xL and their pro-apoptotic counterparts such as Bad, Bax. KGF resulted in variable increases in the expression of the Bcl-2 and Bcl-xL proteins with the exception of Bax and Bad proteins (Figure 6A, B). The combination of KGF and LY was able to decrease Bcl-xL expression which was up-regulated by KGF (Figure 6A). KGF also stimulated the phosphorylation of Bad protein with antibody against p-Bad at Ser-136 in the Western blot analysis in MCF-7 cells (Figure 6B). These results suggested that KGF-induced anti-apoptotic effects in MCF-7 cells involved both pro- and anti-apoptotic proteins and were partially regulated by PI3K/Akt pathway.
Effects of 4-OH-Tam and KGF on the activation of caspase-9 in MCF-7 cells. The involvement of Bcl-2, Bcl-xL, and Bax implied that induction of anti-apoptosis by KGF could be mediated via the mitochondrial pathway. Therefore, we used an antibody that could detect the active-form of caspase-9 protein in MCF-7 cell lysates for Western blot analysis. Our data demonstrated that 4-OH-Tam increased expression of the active form of caspase-9 by 1.7-fold after 2 days of treatment in comparison to the control (Figure 7). The KGF plus 4-OH-Tam treatment reduced the active form of caspase-9 protein to about the same extent as that in the control. The combination of LY with KGF and 4-OH-Tam was able to increase the active form caspase-9 protein which was down-regulated by KGF. Taken together, our data suggested that the inhibitory effects of 4-OH-Tam on cell growth could be through the apoptotic signaling pathway which was likely mediated via the mitochondrial pathway. KGF would appear to act as a survival factor to block formation of the active caspase-9 induced by 4-OH-Tam thus inhibiting apoptosis. This anti-apoptotic pathway triggered by KGF was PI3K/Akt mediated.

DNA fragmentation in MCF-7 cells. A, DNA fragmentation in MCF-7 cells after exposure to three concentrations of 4-OH-Tam for 48 hours in DMEM/F12 medium supplemented with 0.2% bovine serum albumin. B, DNA fragmentation in MCF-7 cells after exposure to 20 ng/ml KGF, 10 μM 4-OH-Tam or LY, or the combination of KGF and 4-OH-Tam, KGF and LY, or KGF plus 4-OH-Tam and LY for 48 hours. The genomic DNAs were isolated from same number of both untreated and treated cells and separated on a 0.8% agarose gel at 90 V for 3 hours. The left lane of each graph is the DNA marker (M).

The effects of KGF on the expression and phosphorylation of anti- and pro-apoptotic proteins. A, KGF up-regulated Bcl-xL and Bcl-2 protein expression through an Akt-mediated pathway. MCF-7 cells were serum starved for 24 hours and treated with KGF at the doses indicated or with the combination of 20 ng/ml of KGF and 10 μM of LY for 48 hours. Western blots of cell extracts were probed using the indicated antibodies (anti-Bcl-xL, -Bcl-2, -Bax, and -actin antibodies). Actin was used as a control for equal loading and transfer. The fold change between treatments was determined by desitometry. Results represent the mean value of three independent experiments ± s.d. Single asterisk represents the significant difference (p<0.05) from the Bcl-xL control group. Double asterisks represent the significant difference (p<0.05) from the Bcl-2 control. B, KGF stimulated p-Bad phosphorylation. MCF-7 cells were serum starved for 24 hours and treated with 1, 5, 10 and 20 ng/ml KGF for 20 minutes. Western blots of cell extracts were probed using the indicated antibodies (anti-Bad, and -p-Bad antibodies). Bad was used as a control for equal loading and transfer. The fold change between treatments was determined by desitometry and results are present as a percentage of that of the control (C). Results represent the mean value of three independent experiments ± s.d. Asterisks represents the significant difference (p<0.05) from the control group.
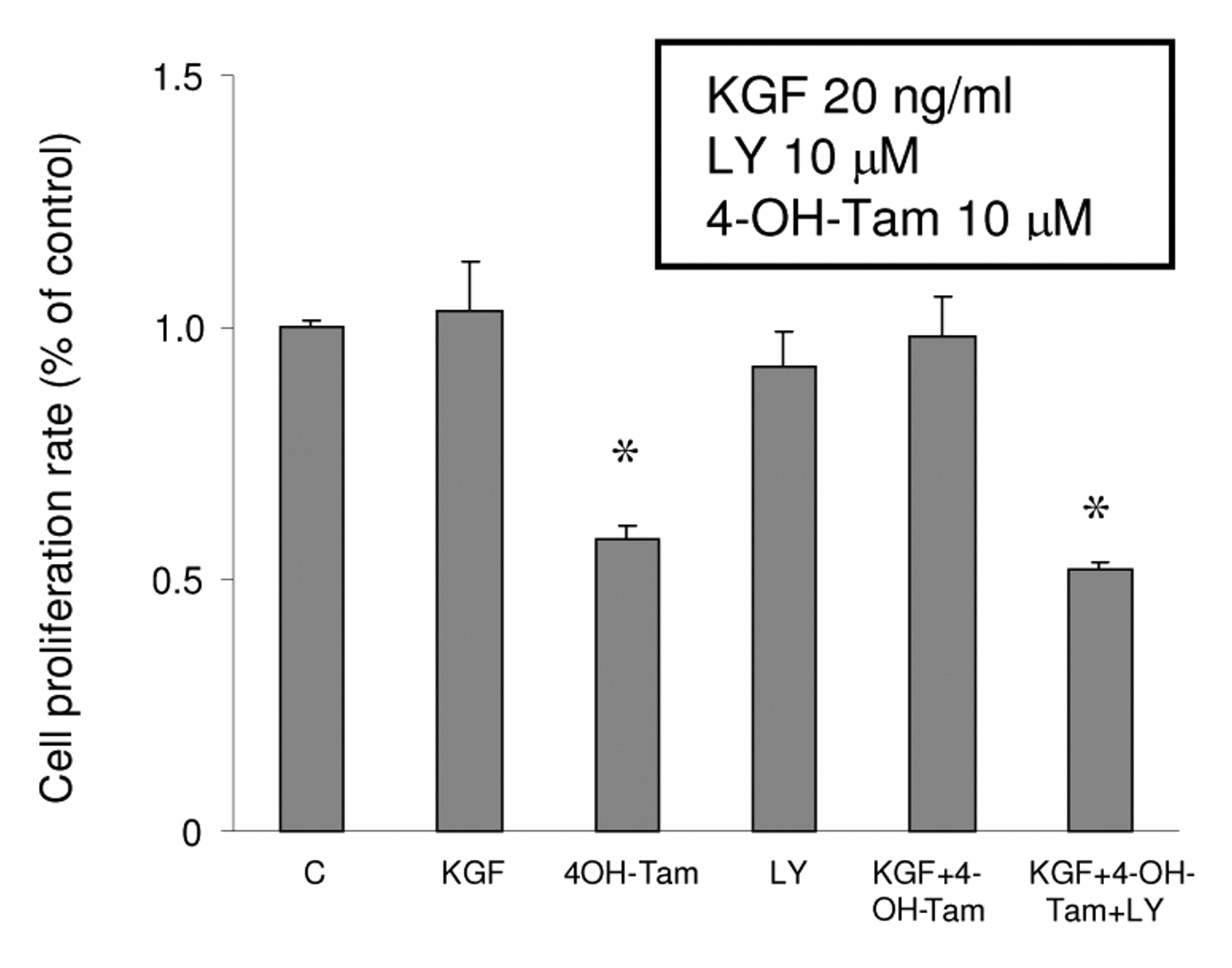
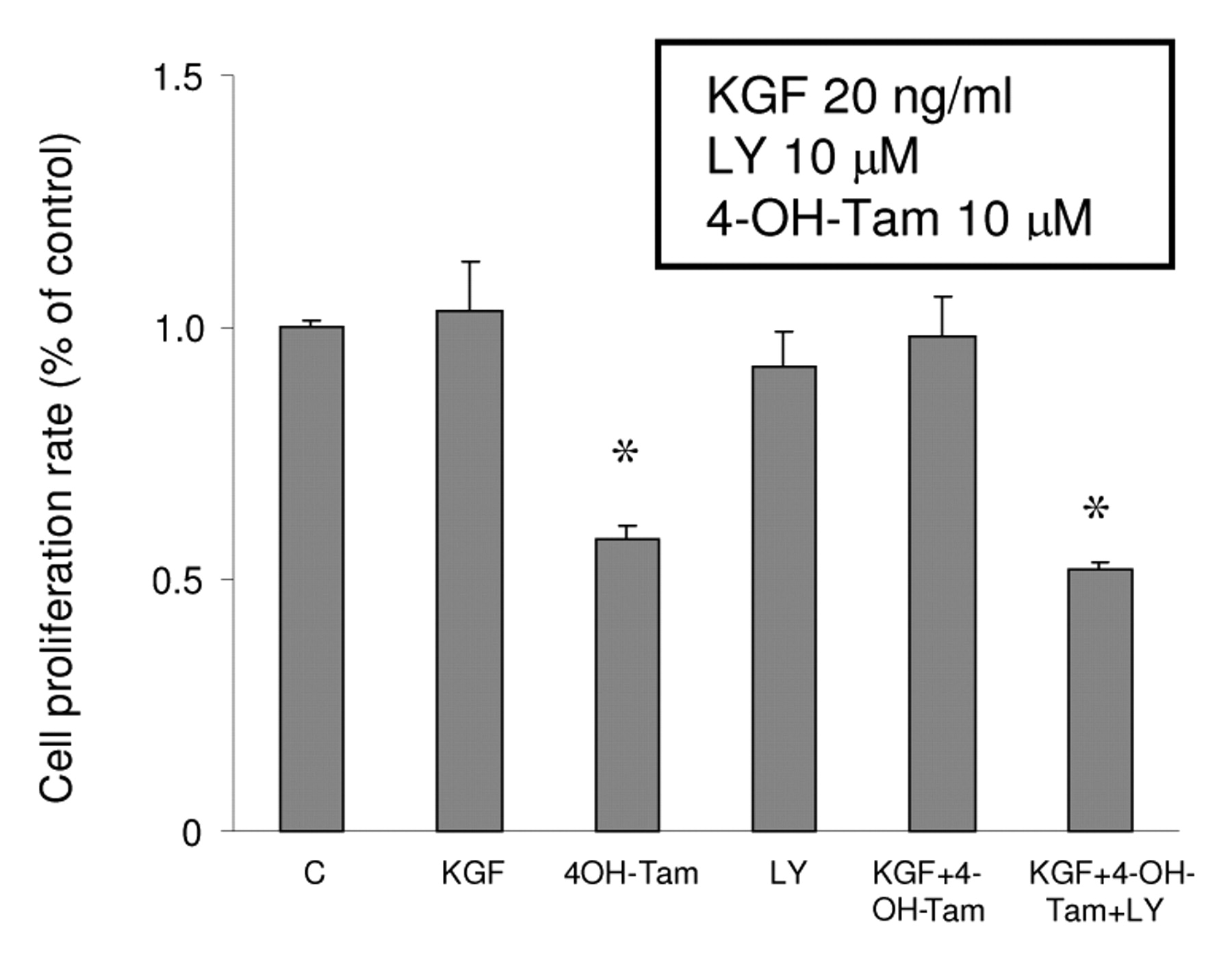
Involvement of PI3K/Akt in the survival effect of KGF. It is interesting to know if the ER-α modulation by KGF has any biological action on MCF-7 cells because the down-regulation of ER-α and anti-apoptotic effects induced by KGF/PI3K/Akt could be the possible mechanism for KGF-induced Tam resistance in breast cancer cells. To demonstrate the role of KGF/PI3K/Akt in the cell survival against 4-OH-Tam in MCF-7 cells, MCF-7 cells were treated by either 20 ng/ml KGF, 10 μM 4-OH-Tam, 10 μM LY, the vehicle, 4-OH-Tam plus KGF, or the combination of 4-OH-Tam, KGF and LY for 3 days. The non-radioactive cell proliferation assay was used in the end of treatments. Figure 8 shows 4-OH-Tam reduced cell proliferation 42% of MCF-7 cells compared to control. KGF alone did not stimulate MCF-7 cell growth, however, the combination of KGF and 4-OH-Tam maintained cell survival at the same level as the control. LY alone did not inhibit cell growth significantly. Our data suggested that KGF disrupted the cell killing effect of 4-OH-Tam and this effect was regulated via the PI3K/Akt pathway.
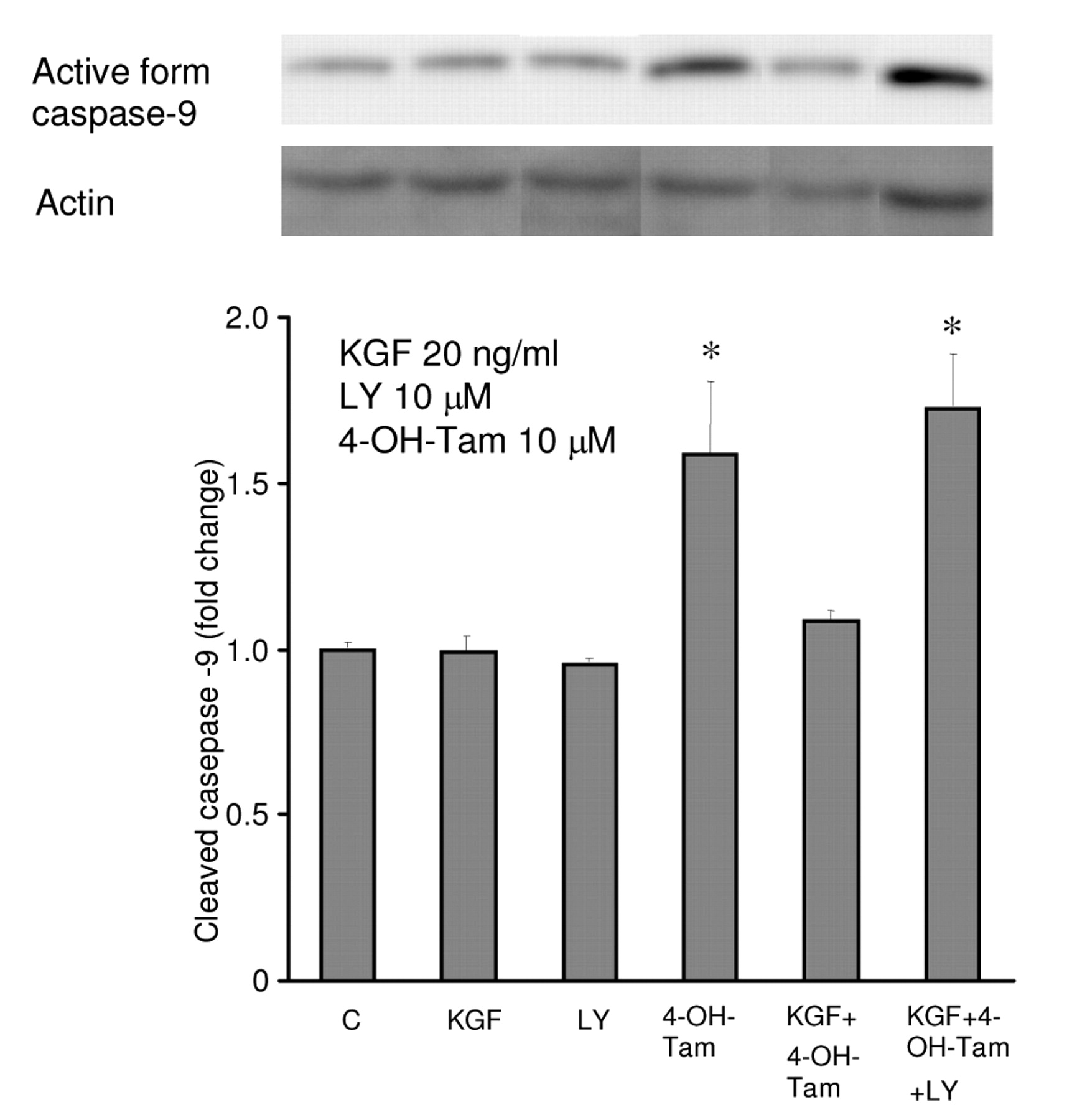
Western blot analysis of the expression of the active form caspase-9 at MW 35 kDa in MCF-7. MCF-7 cells were serum starved for 24 hours and treated with KGF, KGF plus 4-OH-Tam, or the combination of KGF and LY for 48 hours. The doses were as indicated. Western blot of cell extracts were probed using the indicated antibodies (anti-caspase-9 antibody). 4-OH-Tam 10 μM increased expression of the active form of caspase-9 by 1.6-fold respectively after 2 days of treatment. KGF 20 ng/ml inhibited 4-OH-Tam-induced expression of the active form of caspase-9. The PI3K inhibitor LY 10 μM blocked the effect of KGF on 4-OH-Tam treatment. Actin was used as a control for equal loading and transfer. The ratio between the active form of caspase-9 and actin was determined by densitometry and results are represented as a percentage of that of the control (C). Results represent the mean value of three independent experiments ± s.d. Asterisks represent the significant difference (p<0.05) from the control group.
Discussion
The regulation of ER-α expression and apoptosis is a fundamental feature in the progression of human breast cancer. The role of KGF in this process is not well documented. Previously, we reported that KGF down-regulates ER-α expression and up-regulates anti-estrogenic resistance in MCF-7 cells (41). To expand our understanding upon these observations, we focused on the PI3K/Akt signaling axis, a known pathway with protective effects that would inhibit cell apoptosis and affect ER-α expression after being activated. In ours experiments, we have addressed the role of KGF in the model of anti-estrogen-induced apoptosis in hormone-responsive human breast cancer cells. We determined that KGF induces Akt phosphorylation in a dose- and time-dependent manner in MCF-7 cells. We then revealed the involvement of apoptosis in KGF-induced Tam resistance. In particular, KGF increases Bcl-2 and Bcl-xL expression and triggers the phosphorylation of Bad protein. The increase of the active form of caspase-9 protein further confirms that suppression of apoptosis induction of anti-apoptosis by KGF treatment occurs via the mitochondrial pathway. We also determined that the inhibitory effect on apoptosis provided by KGF and the down-regulation of ER-α expression is dependent on activation of the PI3K/Akt pathway. These data support our hypothesis that KGF preserves human breast cancer cell viability by inhibiting apoptosis and probably by adjusting ER-α expression which is PI3K/Akt mediated.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The effect of KGF, LY and 4-OH-Tam on the cell proliferation rate. A total of 1,000 cells/well were seeded in 96-well plates in DMEM/F12 supplemented with 5% FBS overnight. The medium was changed to phenol red-free DMEM/F12 with DCC FBS (1%) and cells were treated with 4-OH-Tam at 10 μM in the presence or absence of either 20 ng/ml KGF or LY 10 μM. Cell proliferation rate was determined by CellTiter™ AQueous assay and optical density was measured at 490 nm by an ELISA plate reader. Results are mean values of four independent experiments and are presented as a percentages related to the control (± s.d.). Asterisks represent significant differences (p<0.05) from the control group.
Tam has been used in the treatment of human breast cancer patients for over a decade. The primary antitumor mechanism of Tam is believed to be through the inhibition of estrogen receptor. Recent studies have implicated the role of caspases in Tam-induced apoptotic signaling. We showed that 4-OH-Tam induced apoptosis in a DNA fragmentation assay and increased the active form of caspase-9 expression in the breast cancer cell line, MCF-7. Caspase-9 is the terminal executor of apoptosis mediated through mitochondria. Thus, our data suggest that 4-OH-Tam-induced apoptosis may be partially through the mitochondrial pathway.
The Bcl-2 family is comprised of both anti-apoptotic and pro-apoptotic proteins which have been established to be upstream of the apoptotic mitochondrial pathway. We observed up-regulation of Bcl-2 and Bcl-xL proteins by KGF. The increase in Bcl-2 and Bcl-xL expression and the decrease in active form caspase-9 might be interpreted as an attempt of KGF to protect the human breast cancer cells from apoptosis through the apoptotic mitochondrial pathway; numerous studies have suggested that the survival or death of human breast cancer cells is determined by an altered balance between pro-apoptotic and anti-apoptotic proteins such as the ratio of Bcl-2 to Bax (43, 44).
The importance of the PI3K/Akt pathway in cell survival and proliferation has been demonstrated in many cell lines (45-48). In our MCF-7 cell model, KGF did not stimulate cell growth. However, KGF maintained cell survival in the presence of 4-OH-Tam. KGF stimulation of the Akt protein phosphorylation is further supported by inhibitor experiments. Our results suggest the predominant role of the PI3K/Akt pathway for the inhibition of anti-estrogenic action triggered by KGF. Focusing further on KGFR/PI3K/Akt kinase signaling, its inhibition by LY resulted in a decrease of Bcl-xL expression and an increase of the expression of the active form of caspase-9. These observations are in agreement with previous reports that Bcl-xL is downstream of PI3K/Akt kinase signaling and that down-regulation of mitochondrial proteins is responsible for the pro-apoptotic response caused by suppression of Akt kinase (49-52).
Bad protein is one of the Akt targets with implication for cell survival regulation. Non-phosphorylated Bad protein will inhibit Bcl-2 and other anti-apoptotic Bcl-2 family by direct binding; thus the phosphorylated form of Bad protein is then localized in the cytosol and its proapoptotic ability is neutralized. The expression of Bad and Bax proteins were not affected by KGF. However, the phosphorylation of Bad protein was triggered by KGF. Our experiments suggest that the phosphorylation of Bad protein is also involved in the survival regulation of MCF-7 cells. The involvement of the PI3K/AKT pathway in the KGF-induced Bad phosphorylation requires further investigated.
Conclusion
In summary, our investigation suggests that KGF exposure may play an important role in the development of resistance to 4OH-Tam in the MCF-7 human breast cancer cell line. Culture of MCF-7 cells under KGF treatment can alter Tam sensitivity by the down-regulation of ER-α and by anti-apoptotic effects through the PI3K/Akt pathway. Therefore, our data support the hypothesis of PI3K/Akt signaling as a mechanism for acquiring resistance to anti-estrogens during human breast cancer progression. Our results here clearly show that new strategies for development and design of novel effective therapeutic drugs based on the inhibition of KGF signaling pathways are needed for treatment of human breast cancer patients.
Acknowledgements
This work was supported in part by NIH grants: CA 94718 and CA 95915 and by Department of Defense (DOD) Breast Cancer Research Programs Grants: DAMD17-99-1-9341 and DAMD 0391.
- Received February 12, 2009.
- Revision received May 18, 2009.
- Accepted June 11, 2009.
- Copyright© 2009 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved