

Abstract
CWC-8 is a new synthesized novel 2-phenyl-4-quinolone compound in our laboratory which has demonstrated potential antitumor activity. In this study, we have defined the viability inhibition and apoptotic mechanisms of CWC-8 on human osteogenic sarcoma U-2 OS cells. According to the MTT assay, the cell viability was inhibited by CWC-8 in a dose- and time-dependent manner, with an IC50 of 4.97±0.24 μM. CWC-8 treatment induced G2/M arrest and apoptosis in U-2 OS cells by cell cycle and flow cytometry analysis. It also profoundly caused a decrease in polymerized tubulin levels by in vitro tubulin polymerization assay which indicated that the microtubular cytoskeleton appears to be one of the cellular targets in response to CWC-8. Western blotting and CDK1 kinase assay showed that CWC-8 treatment caused a time-dependent increase of Cyclin B and CDK1 protein levels and activity during G2/M arrest. CWC-8 treatment also caused a time-dependent increase in Fas/CD95, FADD, cytosolic cytochrome c, caspase-8/-9/-3 active form, Apaf-1, AIF, Bax protein levels, and decrease in Bcl-2 protein level. CWC-8 also promoted caspase-8/-9 and -3 activities; however, pretreatment of cells with pan-caspase, caspase-8/-9 and -3 inhibitors led to reduced cell growth inhibition action. Taken together, these findings show CWC-8 could be a potential candidate for cancer therapy.
- CWC-8
- mitotic arrest
- apoptosis
- human osteogenic sarcoma U-2 OS cells
Human osteogenic sarcoma is a bone tumor that occurs mostly in adolescents and young adults. It is a malignant tumor characterized by the formation of immature bone by the tumor cells (1). Surgery and chemotherapy are the two main methods for clinical therapy (2). Many chemotherapeutic agents are already used, such as methotrexate, doxorubicin, cisplatin, etoposide, isofamide, cyclophosphamide, actinomycin D and Bleomycin (1, 2). However, serious side-effects and drug resistance of these medicines are still problems.
Apoptosis, or programmed cell death, is the well-known phenomenon appearing in cancer cells. Because cancer cells are extremely dependent on aberration of apoptotic pathways to stay alive, focus on apoptosis-inducing agent development for cancer therapy should be a good strategy (3). Two pathways are generally classified: the signal through death receptors (extrinsic pathway) and those involved in the mitochondria (intrinsic pathway). The extrinsic pathway is activated by the engagement of death receptors on the cell surface (3-5). Fas ligand (FasL) to Fas (CD95), tumor necrosis factor (TNF) to TNF receptor (TNFR) and TNF-related apoptosis-inducing ligand (TRAIL) to TRAIL receptors (TRAILRs): death receptor 4, 5 (DR4, DR5) are three major initiative routes of death receptor pathway. The ligand and receptor interactions lead to the recruitment of the Fas-associated death domain (FADD) and the formation of death induced signaling complex (DISC). DISC in turn recruits caspase-8 and promotes procaspase-3 activation. Active caspase-3 causes the cell death phenotype characterized by DNA fragmentation, chromatin condensation, cell shrinkage, membrane blebbing and apoptotic bodies formation. The intrinsic pathway is triggered by various situations, such as growth factor withdrawal, hypoxia, and mitotic defect or DNA damage. Mitochondria are the key organelle to receive and pass down these death stresses. The release of cytosolic cytochrome c, Apaf-1 and pro-caspase-9 from the mitochondria is a particularly important event. This leads to the recruitment of pro-caspase-9 with cytochrome c and Apaf-1 into a large protein complex called the apoptosome. Formation of apoptosome leads to activation of caspase-9 and downstream cascades (i.e. caspase-3/-6/-7) that should cause apoptosis. Some regulatory proteins are from the well-known bcl-2 family. The balance between pro-apoptotic (Bad, Bax, Bid) and anti-apoptotic (Bcl-2, Bcl-xL) proteins of this family decides the outer mitochondrial membrane permeability (6, 7). All these proteins involved in the apoptosis effect are the valuable targets in developing a new drug.
Microtubules are important cytoskeletal components which take part in the regulation of cell proliferation and apoptosis (8, 9) and play a crucial role in many cell functions, such as cell movement, shape, adhesion, signaling and mitosis (10). Microtubules are dynamic cytoskeletal fibers that are composed of α- and β-tubulin heterodimers. There are three main binding sites of microtubule-targeting agents that have been discovered: the paclitaxel site, the Vinca domain and the colchicine domain. Anti-microtubule drugs are generally classed into two main groups by their actions on microtubule polymerization, stabilization or destabilization (11). Cell cycle arrest in G2/M phase transition preceding the apoptosis induction is usually observed from the application of anti-microtubule agents (8, 12). 4-Quinolone, a natural alkaloid and the main skeleton of 2-phenyl-4-quinolone (2PQ), displayed many pharmacological activities, such as antibacterial activity, inhibition of leukotriene biosynthesis and monoamine oxidase activity (13-15). We have reported the biological evaluation of a series of 2PQ class of antimitotic and antitumor compounds. According to National Cancer Institute cell line in vitro screening, some molecules showed promising cytotoxicity effects in the low concentration range (10, 16-18). Unquestionably, 2PQ has the pharmacological potential to be a pioneer of new anticancer drugs. CWC-8 is a newly synthetic agent derived from 2PQ. Through the structure-activity relationship modification from 2PQ, CWC-8 should be a good candidate in cancer chemotherapy. In this study, we used human osteogenic sarcoma U-2 OS cells for examining the induction of cell cycle and apoptosis by CWC-8.
Materials and Methods
Synthesis of CWC-8. Chlorination (Figure 1A): Naphthalene-1-carboxylic acid (998.6 mg, 5.8 mmol) was weighed into a round-bottom flask with 100 ml toluene. A solution of oxalyl chloride (1840.5 mg, 14.5 mmol) was added dropwise. After the mixture was stirred at room temperature for 30 minutes, 2-3 droplets of dimethylformamide (DMF) were added as a catalyst. Over 6 hours later, the mixture was concentrated on a rotary evaporator to remove excess of oxalyl chloride. The semi-oil residue is naphthalene-1-carbonyl chloride. Amide bond formation (Figure 1B): Naphthalene-1-carbonyl chloride (991.3 mg, 5.2 mmol) was dissolved in 200 ml toluene and 4 ml triethylamine. 2-Acetyl-4,5-(methylene-dioxy)aniline (895.9 mg, 5 mmol) was added. The mixture was stirred at room temperature overnight then concentrated on a rotary evaporator to obtain a brown residue. The product was washed with acetone and ethanol several times and dried under vaccum to obtain a yellow solid. Cyclization (Figure 1C): The solid (1333.2 mg, 4 mmol) was suspended in 80 ml of tert-butyl alcohol. A total of 20 mmol potassium tert-butoxide was added and the mixture was heated under reflux for 48 hours. The mixture was cooled and poured into aqueous ammonium chloride solution. The precipitate was collected and washed successively with water, acetone and ethanol. The crude product was purified by flash chromatography which used a mixture of CH2Cl2 and EtOH (16:1-12:1) as eluant.
Cell culture and viability assay. The human osteogenic sarcoma U-2 OS cell line was obtained from the Food Industry Research and Development Institute (Hsinchu, Taiwan, ROC). U-2 OS cells were plated onto 75 cm2 tissue culture flasks with 90% McCoy's 5a medium (Gibco-BRL, Grand Island, NY, USA). All cell media with 1.5 mM L-glutamine were adjusted to contain 1.5 μg/l sodium bicarbonate and supplemented with 10% fetal bovine serum (Gibco BRL), and 1% penicillin-streptomycin (100 Units/ml penicillin and 100 μg/ml streptomycin). Approximately 5×105 cells/well of U-2 OS cells were grown in a 24-well plate for 24, 48 and 72 h at 37°C under a humidified 5% CO2 atmosphere after which time different concentrations of CWC-8 were added (0, 2.5, 5, 7.5 and 10 μM) and cells incubated for a further 48 h. Cells were harvested by centrifugation. Cell viability was determined by the MTT method (19). After incubation for the indicated time, MTT dye was added to each well. After an additional 4 h incubation, the growth medium was removed and the formazan crystals, formed by oxidation of the MTT dye, were dissolved with dimethyl sulfoxide (DMSO) in isopropanol. The absorbance was measured at OD570 and the cell survival ratio was expressed as a percentage of the control (cells with medium alone). For morphological changes, cells were examined and photographed under phase-contrast light microscopy.
Determinations of cell cycle transition and apoptosis by propidium iodide (PI) staining. For cell cycle and apoptosis determination, cells were fixed gently by putting them in 70% ethanol at 4°C overnight and they were then re-suspended in phosphate-buffered saline (PBS) containing 40 μg/ml PI, 0.1 mg/ml RNase and 0.1% Triton® X-100 in the dark for 30 min at 37°C. These cells were then analyzed by flow cytometry (FACS Calibur®; Becton Dickinson, NJ, USA).
In vitro tubulin turbidity assay. Tubulin polymerization was assayed by the use of CytoDYNAMIX Screen 03 kit (Cytoskeleton Inc., Denver, CO, USA) (10). Tubulin proteins (>99% purity, included in this kit) were suspended in G-PEM buffer containing 80 mM PIPES, 2 mM MgCl2, 0.5 mM EDTA, and 1.0 mM GTP (pH=6.9) and 5% glycerol with or without the compound. The mixture was transferred to a 96-well plate and the absorbance was measured at 340 nm (37°C) for 60 min (SpectraMAX Plus; Molecular Devices Inc., Sunnyvale, CA, USA).
Western blotting. Cytosolic fraction and total protein were prepared and determined as previously described (20-21). Equal amounts of (30 μg) proteins were separated by SDS-PAGE and transferred onto PVDF membrane (Millipore, MA, USA). Blots were blocked in PBS buffer (0.05% Triton® X-100 in PBS) containing 5% non-fat milk for 1 h, then the membrane was incubated overnight at 4°C with specific antibodies: cyclin-dependent kinase 1 (CDK1), cyclin B, cyclin A, Fas, Fas-Associated protein with Death Domain (FADD), cytochrome c, apoptotic peptidase activating factor 1 (Apaf-1), caspase-9, caspase-8, caspase-3, apoptosis inducing factor (AIF), Bax, Bcl-2 and β-actin (Santa Cruz Biotechnology, CA, USA). Subsequently, the membrane was washed with phosphate-buffered saline and tween (PBST) buffer and incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies (Santa Cruz Biotechnology, CA, USA). Specific proteins were detected by using Western Blot Chemiluminescence Reagent Plus kits (NEN® Life Science, MA, USA).

Synthesis of CWC-8 from naphthalene-1-carboxylic acid: A, chlorination; B, amide bond formation; C, cyclization.
Caspase-3/-8/-9 activity assay. Approximately 1×107 cells were placed in 75 cm2 flask, 5 μM of CWC-8 were added and cells were incubated for 24 h. Caspase-3/-8 and caspase-9 activity was assessed by caspase colorimetric assay kit (R&D Systems, MN, USA) according to the manufacturer's instruction. Cells were lysed in lysis buffer (50 mM Tris-HCl pH 7.4, 1 mM EDTA, 10 mM EGTA, 10 mM digitonin and 2 mM DTT). Cells mixed with lysis buffer and put on ice, then centrifuged with 13000 rpm and 20 min. Transfer the suspension into new tube. After quantified, fifty μg protein were incubated with caspase-3-, caspase-9- and caspase-8-specific substrates (Ac-DEVD-pNA, Ac-LEHD-pNA and Ac-IETD-pNA; R&D Systems) for 1 h at 37°C. The caspase activity was determined by measuring OD405 of the released pNA (22).
CDK1 kinase assay. CDK1 kinase activity was analyzed according to the protocol of Medical & Biological Laboratories CDK1 kinase assay kit (Medical & Biological Laboratories Co, Ltd, Japan) (22). In brief, the ability of cell extract prepared from each treatment to phosphorylate its specific substrate, MV Peptide, was measured.
Statistical analysis. Our results are expressed as mean ± SD of triplicate samples, and the difference between groups was analyzed by two-tailed Student's t-test, with p<0.05 taken as significant.
Results
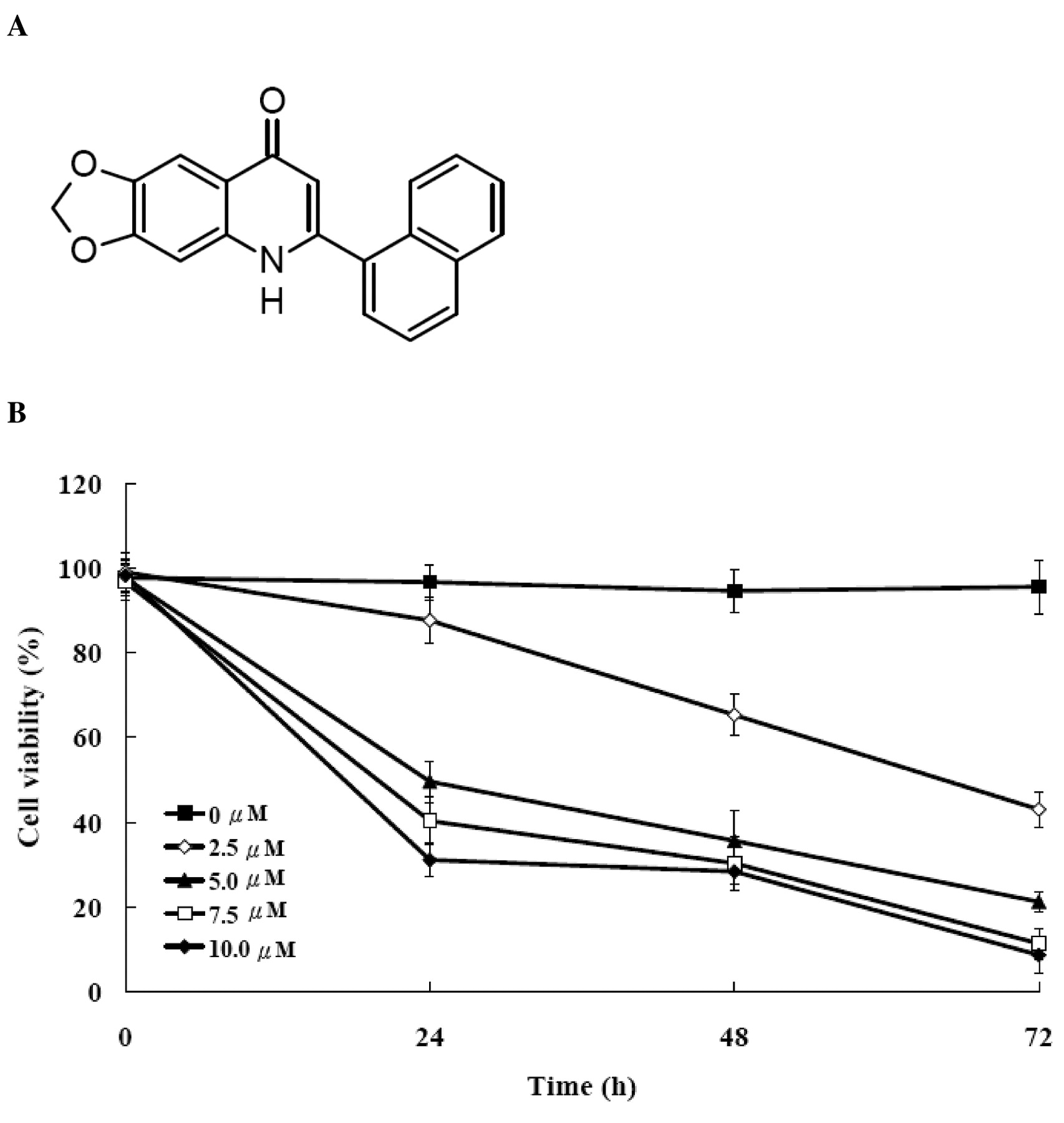
Effect of CWC-8 on cell viability in U-2 OS cells. We determined growth inhibition effects of CWC-8 (Figure 2A) on U-2 OS cell growth. Figure 2B shows the percentage of the cell viability decreased with both the incubation time and CWC-8 concentration. CWC-8 caused dose- and time-dependent inhibition of cell viability, with an IC50 of 4.97±0.24 μM at 24 h.
Effect of CWC-8 on cell-cycle transition and tubulin polymerization in U-2 OS cells. Figure 3A shows that CWC-8 caused an arrest of cell-cycle transition in G2/M phase, and the proportion of cell in the G0/G1 phase declined in a time-dependent manner. The preparation of cells in the G2/M and sub-G1 phases were increased markedly in comparison with these in the G0/G1 phase. These data establish the cytotoxicity efficiency of CWC-8 on U-2 OS cells.
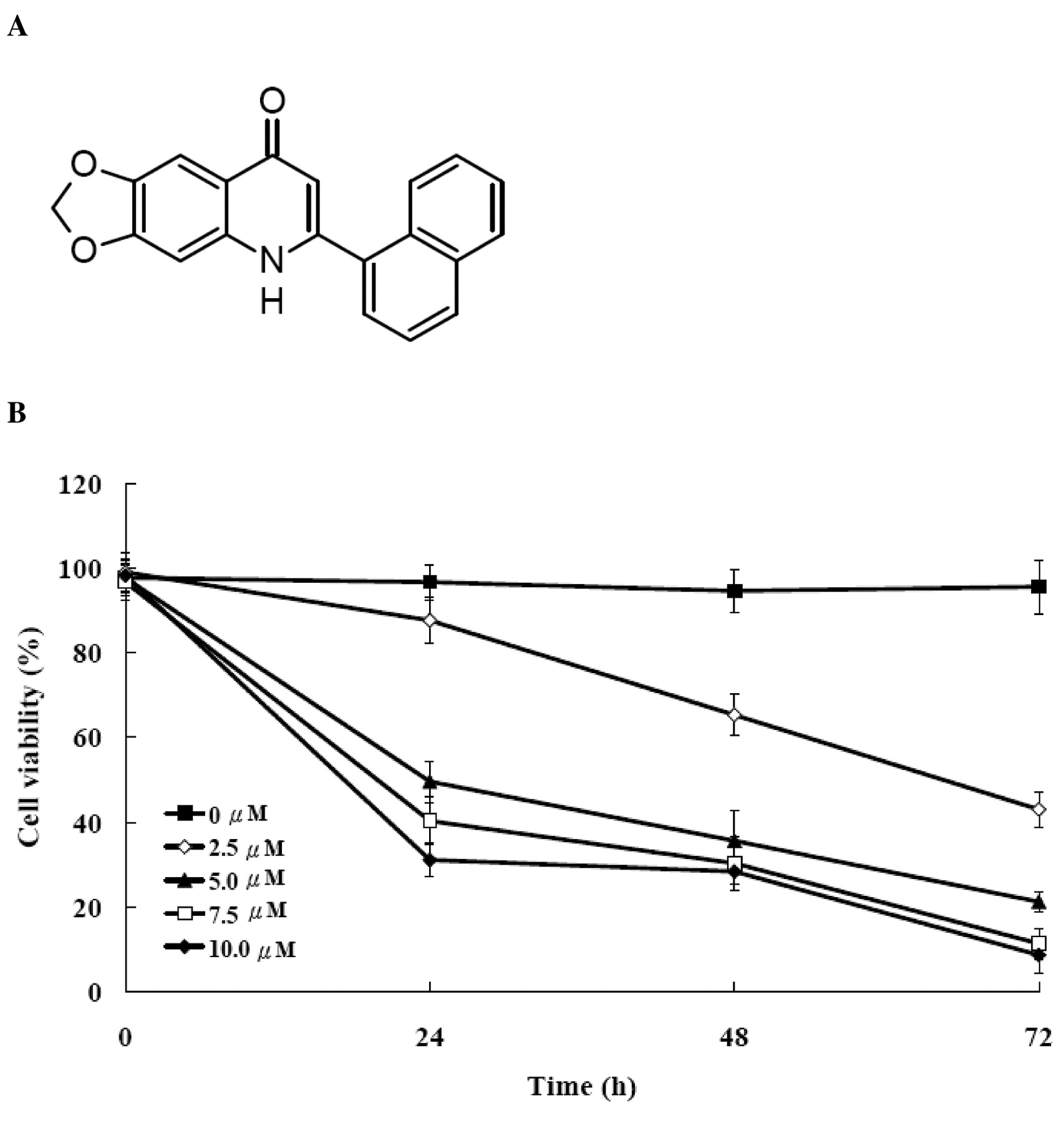
A, Chemical structure of CWC-8 (6-(naphthalen-1-yl)[1,3]dioxolo [4,5-g] quinolin-8(5H)-one) and B, effects of CWC-8 on cell viability in U-2 OS cells. U-2 OS cells were cultured with 0, 2.5, 5, 7.5 and 10 μM of CWC-8 for 24, 48 and 72 h. Percentages of viable cells were determined by MTT assay as described in Materials and Methods. Each point is mean±S.D. of three experiments.
The in vitro turbidity assay of tubulin polymerization was performed to examine if CWC-8 has a direct interaction with tubulin. In Figure 3B, our results demonstrate that the tubulins were assembled in a time-dependent way in the control experiment (DMSO). As well as CWC-8, taxol (a microtubule polymerizing agent) and vincristine (a microtubule de-polymerizing agent) were tested as reference compounds. The result of the reference substances were as expected: taxol boosted, while vincristine suppressed the tubulin polymerization in contrast with the control. As for CWC-8, it had an inhibitory effect on tubulin polymerization in a dose-dependent manner.
Effect of CWC-8 on G2/M phase protein expression and CDK1 activity in U-2 OS cells. Cyclin B/CDK1 complex takes the responsibility to modulate cell-cycle progression from G2 to M phase. Figure 4A shows there was no significant change in Cyclin A level after the treatment with CWC-8. However, the level of Cyclin B and CDK1 expression demonstrated distinct increases with time. CDK1 activity rose dramatically after 6, 12 and 24 h (Figure 4B).

Effect of CWC-8 on cell-cycle distribution (A) and microtubule assembly in vitro (B) in U-2 OS cells. Cells were treated with 5 μM of CWC-8 for 0, 12, 24 and 48 h. Cell cycle analysis was determined by flow cytometric assay as described in Materials and Methods. Each point is mean ± S.D. of three experiments. Tubulin proteins (greater than 99% purity) were suspended in G-PEM buffer plus glycerol in absence (Control) or presence of 1, 5 and 10 μM of CWC-8, 10 μM taxol or 10 μM vincristine at 4°C. Absorbance was detected each minute for 60 minutes at 340 nm at 37°C.

CWC-8 affects the protein levels (A) and CDK1 activity (B) of U-2 OS cells. Cells were treated with 5 μM of CWC-8 for 0, 6, 12 and 24 h then the total proteins were prepared then detected by Western blotting and CDK1 activity. Primary antibodies for CDK1, cyclin A, cyclin B were examined by Western blotting. CDK1 kinase activity was measured in cellular extracts for the ability to phosphorylate MV Peptide, a CDK1 kinase-specific substrate, according to Medical & Biological Laboratory's CDK1 kinase assay kit.
Effect of CWC-8 on apoptosis related protein expression in U-2 OS cells. Figure 5A shows that CWC-8 caused apoptosis in U-2 OS under microscopy. We investigated the change in protein expression of Fas, FADD, cytochrome c, Apaf-1, AIF, Bcl-2, Bax, active caspase-8, -9 and -3 after CWC-8 treatment. The results are shown in Figure 5B and 5C. Up-regulated expression proteins included Fas, FADD, cytosolic cytochrome c, cytosolic Apaf-1, cytosolic AIF, Bax, caspase-8, -9 and -3, while Bcl-2 was down-regulated.
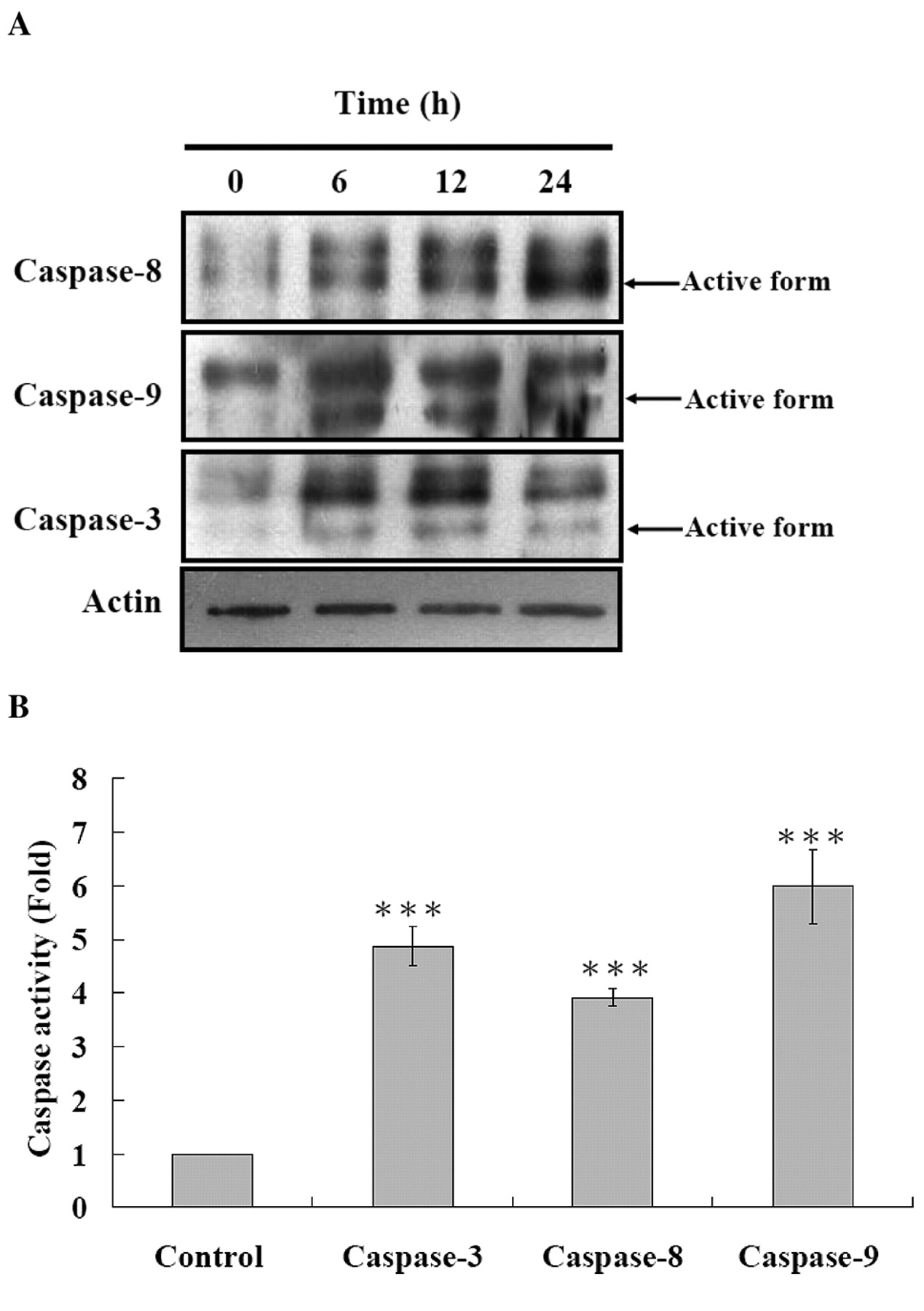
Effects of CWC-8 on the activity of caspase-8, -9 and -3 in U-2 OS cells. We determined caspase-8, -9, -3 activities after 24 h CWC-8 treatment. In Figure 6A, B, CWC-8 induced caspase-8, -9, -3 activities in U-2 OS cells. In order to confirm that CWC-8-induced apoptosis in U-2 OS cells through caspase-dependent pathways, we pretreated cells with caspase-3, -8, -9 and pan-caspase inhibitors. These results are shown in Figure 7. Caspase-3, -8, -9 and pan-caspase inhibitors significantly increased the number of viable cells.
Discussion
According to the pharmacological character of the 2PQ series of compounds, they are a type of antimitotic agent. These molecules displayed outstanding activity of anti-proliferation in various cancer cell lines (16-18). The difference in comparing with other anti-microtubule agents (vincristine, vinblastine, docetaxel and taxol) is that 2PQ also displayed efficiency in inhibiting NCI/ADR-RES cells which exhibit drug-resistance due to their P-glycoprotein-rich expression (23, 24). It is such an advantage to develop these 2PQ agents with this characteristic. CWC-8, a compound derived from 2PQ, revealed this promising activity as expected in our experiments, which has proven mitotic arrest, anti-tubulin polymerization and apoptosis effects in U-2 OS cells.
Apoptosis is the phenomenon observed in CWC-8 treated U-2 OS cells. However, CWC-8 is a type of anti-microtubule compound that arrests cells in G2/M transition. From Figure 3A, we can see that the sub-G1 phase (apoptosis) increase followed the increase in the G2/M population. Much research demonstrated that an anti-tubulin agent leading to the arrest of the cell-cycle in the G2/M phase would induce apoptosis occurrence by a mitochondrial pathway. There are numerous initiating events that have been found, such as activation of the pro-apoptotic Bax and Bad, or in-activation of the anti-apoptotic Bcl-2 by phosphorylation, which was implicated with various kinases like CDK1, JNK, c-Raf, ERK1/2, cAMP-dependent PKA and PKCa (12, 25-27). It is suggested that CDK1 plays a crucial role in inducing the apoptosis signal pathway by anti-microtubule agents. Not only Bcl-2 phosphorylation, but also CDK1 activation was shown to phosphorylate pro-death BH3-only protein Bad on serine 128 (26), which promotes the activation effect of Bad. In this kind of unbalance triggered by CDK1 between pro- and anti-apoptotic proteins, the formation of a permeability transition (PT) pore on the mitochondria membrane and the subsequent releasing of apoptotic factors will occur. We focused on the expression change of Bcl-2, Bax and CDK1 in the CWC-8 experiment. The up-regulated level of Bax, CDK1, CDK1 activity and down-regulated Bcl-2 were consistent. This explains why CWC-8 possesses the apoptotic potency in the intrinsic pathway in U-2 OS cells.

Effects of CWC-8 on cell morphology (A) and the level of apoptosis-associated proteins in U-2 OS cells (B). Cells were cultured with 5 μM of CWC-8 for 24 h. The cells were examined and photographed under phase-contrast microscopy (x400). For western blotting, cells were treated with 5 μM of CWC-8 for various time periods and were then harvested for cytosolic protein determination, as described in Materials and Methods.
In many studies of mitotic modulators, it is usual to see that G2/M arrest was induced by anti-microtubule agents, which leads to mitochondrial permeability transition and then triggering of the downstream cascades of the intrinsic apoptotic pathway. CWC-8 exhibited the activation of extrinsic apoptosis pathway in this study, including up-regulation of expression of Fas, FADD and active caspase-8. In a previous study of 2PQ, only caspase-8 activation but not death receptor level promotion was observed and it was suggested that the extrinsic apoptosis pathway did not play a crucial role in 2PQ action (10). The study indicated that CD95 mediated apoptosis can be disrupted by Bcl-2 and Bcl-XL proteins, which are the blockage proteins implicated in the intrinsic apoptosis pathway (28).

Effects of CWC-8 on the levels change of caspase-8/-9 and -3 proteins (A) and activity (B) in U-2 OS cells. U-2 OS cells were cultured with 5 μM of CWC-8 for 0, 6, 12 and 24 h, and were then harvested for cytosolic protein determination, as described in Materials and Methods. The caspase-8/-9 and -3 protein expressions and activity were estimated by Western blotting and caspase activity assay as described in Materials and Methods; ***p<0.001.
For the extrinsic apoptosis pathway (CD95 signaling pathway), two distinct prototypic cell types have been identified (7, 28, 29). In type I cells, activation of caspase-8 is sufficient to directly activate the downstream caspase-3, leading to apoptosis through the internalization process (3, 30). Activation in type I cells is independent of mitochondria and could not be blocked by anti-apoptotic Bcl-2 family proteins. In type II cells, the amounts of activated caspase-8 are not enough to trigger the activation of caspase-3, hence a mitochondrial amplification loop is required for strengthening the inadequate signal by cleaving pro-apoptotic Bid into its truncated Bid (tBid) through caspase-8. Consequently, tBID binds to Bax and Bak and causes the release of cytochrome c and other pro-apoptotic molecules to pass down the apoptosis signal. Cross-talk between the extrinsic and intrinsic apoptotic pathways is mediated by Bid. Therefore, we may make a reasonable speculation that the U-2 OS cell line is one of the type II cells, and the apoptotic effect of CWC-8 on U-2 OS cells could be blocked by all mitochondria-related anti-apoptotic agents. In past apoptosis transduction studies of the death receptors, MKN45, 293T, SKW6.4, H9 and ACHN were classified as type I cells. Comparatively, HCT116, HCT15, A549, SW480, CEM and Jurkat were identified as type II cells (28, 31, 32).

Caspase-8, -9, -3 and pan-caspase inhibitors inhibited CWC-8-induced growth inhibition in U-2 OS cells. Cells were pretreated with caspase-3 inhibitor (Z-DEVD-FMK) (A), a caspase-8 inhibitor (Z-IETD-FMK) (B), a caspase-9 inhibitor (Z-LEHD-FMK) (C), or pan-caspase inhibitor (Z-VAD-FMK) (D) then cells were treated with 5 μM of CWC-8 for 24 h. U-2 OS cells were harvested for determination of cell viability as described in Materials and Methods. Data represent mean ± S.D. of three experiments; ***p<0.001.
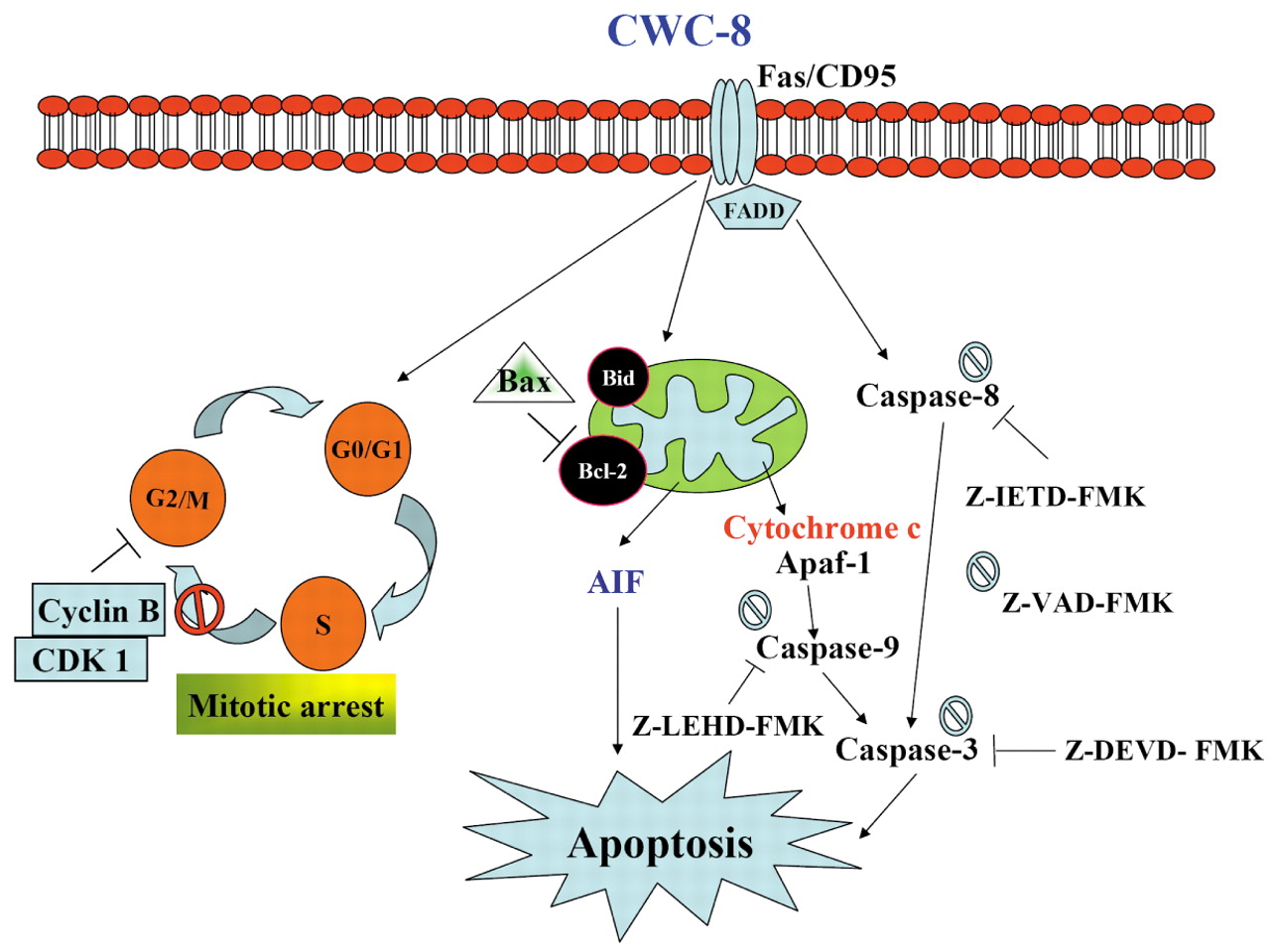
Taken together, our results demonstrated that CWC-8 causes anti-tubulin polymerization and G2/M arrest, and has extrinsic apoptosis and intrinsic apoptosis activities in U-2 OS cells. The proposed molecular mechanism and pathway involved in CWC-8-induced mitotic arrest and apoptosis in U-2 OS cells is outlined in Figure 8. CWC-8 could be a potential candidate for cancer therapy, especially in cases of drug resistance development by the malignant cells. Furthermore, the advanced structure modification of CWC-8 may enable us to conquer these deathful diseases in the future.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The proposed model of CWC-8-mediated mitotic arrest and apoptosis in U-2 OS cells.
Acknowledgements
This investigation was supported by research grants from the National Science Council of the Republic of China (NSC 96-2323-B-039-001; NSC 97-2323-B-039-001) and grants from the China Medical University (CMU 97-238).
- Received February 20, 2009.
- Revision received April 24, 2009.
- Accepted June 10, 2009.
- Copyright© 2009 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved