


Abstract
Background/Aim: Several clinical trials have investigated homologous recombination deficiency and BRCA1/2 status to select ovarian cancer patients for treatment with poly(ADP-ribose) polymerase-inhibitors (PARPi), but less attention has been given to other DNA-damage response (DDR) pathways. Therefore, we investigated somatic single/multiple nucleotide variants and small insertions/deletions in exonic and splice-site regions of 356 DDR genes to examine whether genes other than BRCA1/2 are altered. Materials and Methods: Whole-exome sequencing data from eight high-grade serous adenocarcinoma (HGSC) and four clear cell carcinoma (oCCC) patients were analyzed. Results: Forty-two variants (pathogenic, likely pathogenic or variants of uncertain significance) in 28 genes from DDR pathways were identified. Seven out of nine TP53 variants were previously described in The Cancer Genome Atlas Ovarian Cancer; other variants were found in 23 out of 28 unique genes, whereas no variants were reported in FAAP24, GTF2H4, POLE4, RPA3, and XRCC4. Conclusion: As the identified variants were not only limited to well-known TP53, BRCA1/2, and HR-associated genes, our study might contribute to the better understanding of which DDR pathways potentially influence disease progression. Moreover, they may display a potential role as biomarkers to predict platinum-based chemotherapy or PARPi treatment response or disease progression, as differences in disrupted DDR pathways were observed between patients with long and short overall survival in HGSC and oCCC groups.
Epithelial ovarian cancer (EOC), which represents approximately 90% of ovarian cancer (OC) cases, is diagnosed in late, more lethal, stages of the disease (1, 2). It can be further divided into five main classes: high-grade serous (HGSC, 70%), clear cell (oCCC, 10%), endometrioid (10%), mucinous (3%), and low-grade serous carcinomas (<5%) (3). The 5-year survival rate of late stage EOC patients [International Federation of Gynecology and Obstetrics (FIGO) stage IV] is below 30%, whereas that of patients in early stage (FIGO stage I) is above 90% based on the report from Danish Gynecologic Cancer Database (DGCD) (4). A comprehensive molecular characterization of HGSCs has revealed mutations in several genes: most notably in the TP53 gene, which displayed mutations in almost 96% cases (5, 6). Moreover, it was shown that about 50% of HGSC patients have alterations in the homologous recombination (HR) DNA damage response pathway (7). Germline and somatic BRCA1 and BRCA2 mutations were detected in up to 25% and 3-7% cases of HGSCs, respectively (8). BRCA1 and BRCA2 are essential proteins in the DNA damage response machinery that senses, signals, and repairs DNA lesions (9). Three protein complexes: the MRE11-Rad50-NBS1 (MRN), RPA/ATRIP, and KU70-KU80 are among the first sensors and responders to DNA damage (10). They recruit DNA damage signalling components: ATM, ATR, and DNA-PKs, which triggers repair signalling by phosphorylating key proteins such as BRCA1, CHK1, CHK2, H2AX, p53, and RAD17 (11). Various DNA repair mechanisms are initialized in response to activated signaling-transduction pathways: base excision repair (BER), Fanconi Anemia (FA), HR, mismatch repair (MMR), nucleotide excision repair (NER), and non-homologous end joining (NHEJ) (12, 13). Dysregulation of the DDR may impair genomic instability in cells and promote cancer development, but also may offer targetable weaknesses that can be further exploited therapeutically (14). Defective DDR is an attractive target in EOC, as demonstrated by favorable responses to both platinum chemotherapy and poly (ADP-ribose) polymerase inhibitors (PARPi), which utilize deficiencies in DDR pathways to lead to cancer cell death (7, 12, 15). Although initially the majority of HGSCs are platinum-sensitive, many patients will relapse eventually as platinum-resistant after frequent relapse-response history (12). Resistance to platinum therapy is strongly predictive of resistance to PARPi treatment (16). Olaparib was the first PARPi that received regulatory approval in Europe to treat HGSC patients with a germline (g) or somatic (s) BRCA1/2 mutation (17). Clinical studies have provided evidence of favorable response to PARPi treatment in patients harboring pathogenic/likely pathogenic BRCA1/2 mutations with complete or partial response to platinum-based chemotherapy as compared to patients with no BRCA1/2 mutations (18). Further effort is needed to determine which non-BRCA1/2 patients benefit from such treatment, however, there is yet no consensus reached on how to identify predictors of PARPi sensitivity in OC (19). There is growing evidence that mutations in non-BRCA DDR genes might contribute to better understanding of cancer predisposition and guide the potential treatment of HGSCs by predicting sensitivity to PARPi (20–22). In contrast to HGSCs, oCCCs usually express wild-type p53 protein and have a lower frequency of HR gene mutations (23). The most frequent variants in oCCC are in ARID1A and PIK3CA, both appearing as single or double hit mutations in approximately 50% of patients (23, 24). Moreover, oCCC exhibits higher risk of resistance to platinum-based chemotherapy, which in consequence might lead to poor prognosis (23, 25). Understanding of the DDR mechanisms and their defects may help to target individualized treatment to various subtypes of EOC (26).
We examined the occurrence of somatic single and multiple nucleotide variants (SNV and MNV), as well as small insertions/deletions (INDELs) in exonic and splice site regions of 356 DDR genes in HGSC and oCCC patients using whole exome sequencing. The aim of this study was to investigate whether DDR pathways other than HR may be affected and if differences between HGSC and oCCC groups can be observed.
Materials and Methods
Patient cohort. Fresh-frozen samples were acquired from two Danish projects: the Pelvic Mass study (2004-2014) and the Gynecological Ovarian Vulva Endometrial Cervix cancer (GOVEC) study (2015 – ongoing) through the Bio- and Genome Bank Denmark (27). The study was performed according to the guidelines of the Declaration of Helsinki, including written informed consent from all patients. The study has been approved by the Danish National Committee for Research Ethics, Capital Region (H-17029749/H-15020061). Both patients’ groups: oCCC and HGSC had similar age characteristics: 60.7 (57.2-68.0) and 63.8 (53.8-68.3) years, respectively. Moreover, HGSC patients, were matched on FIGO stage, but their overall survival (OS) differed significantly:“HGSC_ShortOS” with median OS=26.9 months and “HGSC_LongOS”, median OS=131.1 months) (p<0.05, the unpaired two-samples Wilcoxon test) (Table I). Hematoxylin and eosin (H&E)-stained tissue slides neighboring the excised tumor were examined by a trained pathologist to determine percentage of tumor cells.
Patients included in this study, chosen from the Danish Pelvic Mass/GOVEC study.
Whole exome sequencing. Genomic DNA was extracted using Maxwell RSC Tissue DNA (AS1610, Promega, Madison, WI, USA). DNA concentration measurements were performed on a Qubit system with the High Sensitivity dsDNA assay kit (Q33120, Thermo Fisher Scientific, Waltham, MA, USA). Exome sequencing libraries were prepared from 100 ng DNA using the Ion AmpliSeq Exome RDY kit (A38262, Thermo Fisher Scientific) according to the manufacturer’s protocol. Equal amounts of amplified DNA libraries were loaded onto an Ion 550 Chip (A34537, Thermo Fisher Scientific) using the Ion Chef System (Thermo Fisher Scientific). Sequencing was performed on an Ion S5XL System (Thermo Fisher Scientific).
Data processing and variant calling. Sequencing data were acquired, pre-processed, aligned, and analyzed by using Ion Suite Software v5.6 (Thermo Fisher Scientific), coupled with AmpliSeq Exome single sample (Somatic) analysis module. Further filtering for true variants was performed using R environment (28), as described below, and presented in Table II. Only mutations occurring in exonic or splice-site (located within the first 3 nucleotides of the 5′ or 3′ end) regions of the 356 DDR genes were selected. As we expected to find TP53 gene variants in more than 90% of HGSC cases based on previous reports (5), we performed first manual TP53 variant check of non-filtered data from the subjects with HGSC to determine cut-off for a coverage filter (value=49).
Overview of the workflow applied for variant calling.
The following thresholds define applied exclusion criteria for each variant: Coverage <49; Coverage <10% of median coverage; Phred score <100; Ion Reporter™ p-value >0.01; Allele ratio <25% of average allele ratio per sample; Potential germline (allele ratio on target allele=1); Homopolymer >5; Synonymous variant; Strand bias score >60; UCSC Common SNPs; Minor allele frequency >0.01.
All mutations that passed the above-described criteria were summarized, cross-referenced, and clinically annotated using the ClinVar and Varsome databases (data status check: December 01, 2022). Variants classified as “Benign” or “Likely Benign” by the ClinVar and/or Varsome databases were excluded from further analysis. We excluded potential germline variants based on “1000 Genomes” and “GnomAD/ExAc” databases (29, 30). Moreover, the variants were checked in a Catalogue of Somatic Mutations in Cancer (COSMIC) (31). We manually assessed the remaining variants for sequencing and annotation errors with integrated genomic viewer (IGV) (Broad Institute, Cambridge, MA, USA) to confirm or exclude the variant. Furthermore, we investigated possible variants in two genes: ARID1A and PIK3CA, which are the most frequently mutated genes in oCCC (both occur in approximately 50% of patients) with identical thresholds as described above.
External datasets. We collected exome sequencing data from 436 OC patients data from The Cancer Genome Atlas (TCGA) and for detailed information regarding biospecimen collection, clinical data, and sample processing, we refer to the original publications (5). The data were processed by the R package ”Maftools” (32). Moreover, we downloaded the data from the International Agency for Research on Cancer (IARC TP53) database (version R20) (33) to check whether TP53 mutations found in our study had been previously reported.
Assignment of genes to DDR pathways. We included 356 genes belonging not only to the major repair pathways: BER, FA, HR, MMR, NER, and NHEJ, but also involved in main signalling pathways: ATM, ATR, RB, and TP53 pathways (9, 11-13, 34, 35), as described in Table III. Almost all pathways were collected from The Molecular Signatures Database (MSigDB) v7.4 from three databases: Kyoto Encyclopedia of Genes and Genomes (KEGG) (36), Reactome (37), and Biocarta (38) by use of R environment (28). As the FA pathway from KEGG was not available through MSigDB, we downloaded its content directly from KEGG server. As the same pathways from different resources contained different genes, we compiled them into one, e.g., BER pathway consists of all genes presented in KEGG_BASE_ EXCISION _REPAIR and REACTOME_BASE_EXCISION_REPAIR. Each gene has been assigned to one or more pathways, as different pathways share their components.
The full list of the pathways with their accession numbers.
Results
We analyzed 4 oCCC and 8 HGSC samples by exome sequencing with Ion AmpliSeq Exome RDY kit from Thermo Fisher Scientific. Patients’ age characteristics were similar in oCCC and HGSC groups. Moreover, individuals in HGSC group were matched on FIGO stage, but divided into two groups with short and long OS, respectively (Table I). All patients underwent standard first-line combination platinum and paclitaxel chemotherapy. None of the patients received PARP-inhibitor treatment. Treatment-free interval (time from end of first-line to start of second-line therapy or to the date of last follow-up (no later than August 22, 2022) is presented for each patient.
Sequencing data analyzed (n=778,623 variants) were subjected to SNV, MNV, and INDELs variant calling in exonic or splice-site regions of 356 DDR genes, which resulted in n=4,723 variants (Table II). These variants were subjected to further filtering and inspected with IGV to approve or reject them from the final list. Nine different TP53 variants were found in all serous patients and in one of the oCCC samples. All of these variants were previously reported in the IARC database, whereas seven of them were found in a TCGA dataset (c.97-1G>A, c.413C>T, c.646G>A, c.659A>C, c.701A>G, c.731G>A, c.772G>A).
In addition to TP53, we identified 33 variants in a total of 27 DDR genes, which were classified as pathogenic/likely pathogenic (P/LP) (Table IV) or as variants of uncertain significance (VUS) (Table V). All the 33 variants were unique, but some overlap between genes carrying these variants was observed (Figure 1). None of these variants were found in the TCGA cohort, however, other variants were found in 22 genes (ATM, BLM, BRCA1, BRCA2, CDK12, CDK13, CHEK2, COPS6, DDB1, DNA2, ELOA, ERCC4, FANCM, MUTYH, NELFE, POLD1, POLD3, POLR2B, RB1, RFC1, TFPT, TOPBP1) (Figure 2). There were no reported variants in FAAP24, GTF2H4, POLE4, RPA3, and XRCC4.
Summary of pathogenic (P) or likely-pathogenic (LP) somatic variants found in exonic and splice-site regions of DNA-damage response genes by exome sequencing.
Summary of unknown significance variants found in exonic and splice-site regions of DNA-damage response genes by exome sequencing.
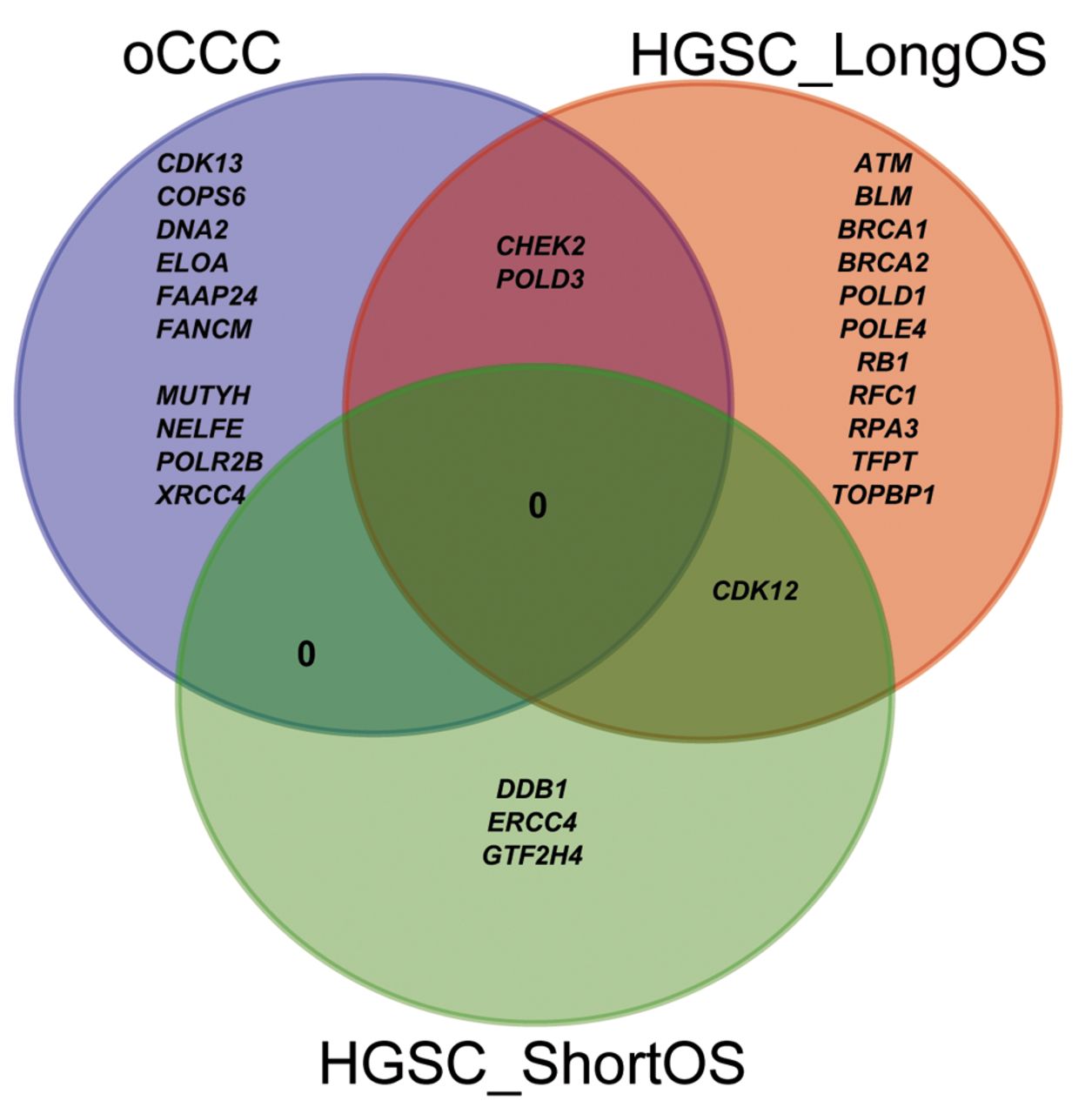
Venn diagram of overlapping genes carrying a variant among the three groups of patients: HGSC_LongOS: High-grade serous carcinoma patient with long overall survival; HGSC_ShortOS: high-grade serous carcinoma patient with short overall survival; oCCC: ovarian clear cell carcinoma.
Moreover, when investigating possible mutations in ARID1A and PIK3CA genes, pathogenic, likely-pathogenic or VUS variants were found exclusively in oCCC patients: PIK3CA: c.3140A>G (P) in oCCC_01, two VUS variants in oCCC_02: ARID1A: c.1780delC and PIK3CA: c.344_346delGAG, and one VUS variant in oCCC_03: ARID1A:c.3428_3429delGG, c.3429delG.
Discussion
Many clinical trials investigated BRCA1/2 variants and/or homologous recombination deficiency (HRD) status, as crucial criteria to select EOC patients with complete or partial response to platinum-based chemotherapy for PARPi treatment (7, 39). Less attention has been given to other DDR pathways in predicting response to treatment (26). In this study, we examined mutations in the exonic and splice-site regions of a total of 356 DDR genes in the same workflow to determine whether DDR pathways other than HR are affected in HGSC or oCCC.
BRCA1/2 were found only in HGSC_LongOS patients, which is in agreement with previous studies that demonstrated prolonged OS with BRCAness phenotype (40, 41). Twelve out of 16 variants (P+VUS) (75%) found in HGSC_LongOS were located in genes that have implications in the HR-pathway. Moreover, each patient in the HGSC_LongOS group carried a variant in a HR-related gene in contrast to HGSC_ShortOS patients (only one patient with HR-deficiency) and oCCC (3 out of 4 patients with HR-deficiency with 3 HR-related variants: 3 out of 13, 23%). It seems that genes that were defective in the HGSC_LongOS group are involved in different DDR pathways (Table IV and Table V, column “SUM”), whereas in HGSC_ShortOS and oCCC they are usually members of only one of the DDR pathways.
None of the 33 variants identified in this study were found in the TCGA cohort that contains only HGSC patients; however, other variants were found in 22 out of 27 unique genes: ATM, BLM, BRCA1, BRCA2, CDK12, CDK13, CHEK2, COPS6, DDB1, DNA2, ELOA, ERCC4, FANCM, MUTYH, NELFE, POLD1, POLD3, POLR2B, RB1, RFC1, TFPT, and TOPBP1 (Figure 2). There were no reported variants in the TCGA cohort for FAAP24, GTF2H4, POLE4, RPA3, and XRCC4 genes. Variants in two of these five non-TCGA genes FAAP24 and XRCC4, were observed only in the oCCC group, but not in the HGSC cohort from TCGA. Further exploration may reveal whether these genes are of special importance for the oCCC group and might be helpful in guiding treatment of oCCC patients. The FANCM/FAAP24 complex detects interstrand crosslinks and recruits other components of the FA core complex (42). Cells with FA deficiency display hypersensitivity to DNA cross-linking agents (43) and it might explain the prolonged OS of the oCCC_03 patient with FAAP24:c.635C>T variant. This patient is also a carrier of DNA2: c.68C>T. Loss of XRCC4 might activate to a back-up repair pathway called alternative end-joining repair (a-EJ) in human somatic cells (44). The a-EJ pathway is considered as a promising therapeutic target in HR or NHEJ deficient cancers, as its inhibition causes death of cells dependent on the a-EJ pathway to repair their DNA, but not normal cells (45). GTF2H4, POLE4, and RPA3 have also been previously reported in cancer and OC studies. Other variants in GTF2H4 have previously been shown to confer susceptibility to lung cancer (46, 47). Interestingly, loss of POLE4 was shown to sensitize HeLa cells to PARPi treatment (48) and hypersensitivity to ATR inhibition (49). Inhibition of RPA:RAD52 protein-protein interactions was reported to cause selective death of HRD cancer cell lines (50). Moreover, RPA exhaustion was reported to determine cisplatin response in HGSC cells (51).
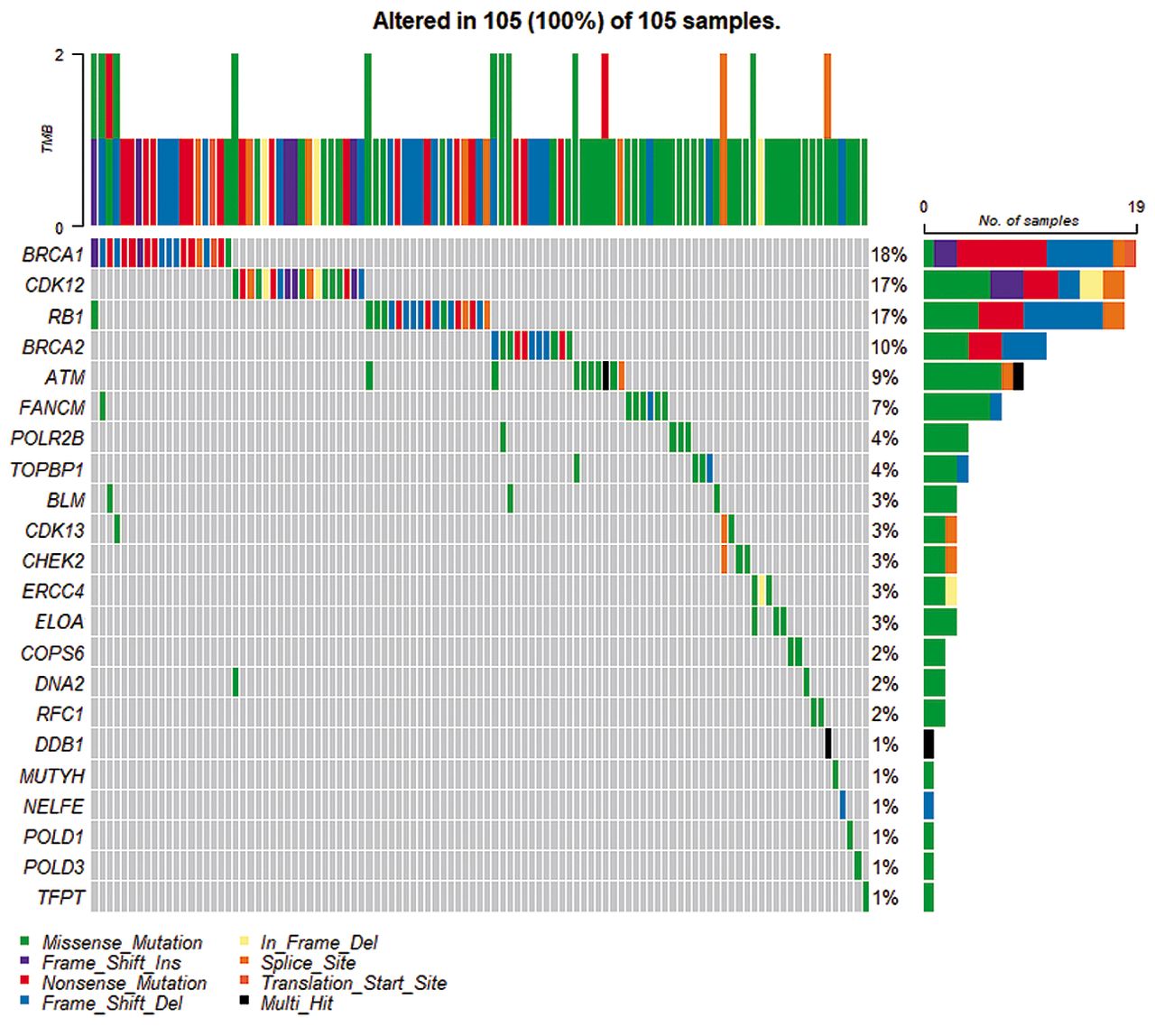
DNA-damage response genes, in which we found 33 somatic variants. were submitted as a query to The Cancer Genome Atlas Ovarian Cancer dataset to find if any single/multiple nucleotide variants and small insertions/deletions variants for those genes have been previously reported. Missense_Mutation: The point mutation alters the protein structure by one amino acid; Nonsense_Mutation: a premature stop codon is created by the variant; Frame_Shift_Ins: insertion that moves the coding sequence out of frame; Frame_Shift_Del: deletion that moves the coding sequence out of frame; In_Frame_Del: deletion that keeps the sequence in frame; Splice_Site: the variant is within a configurable number of bases of a splice site; Multi_Hit: more than one variant type.
TP53 SNVs were found in all patients with HGSC, whereas variants in ARID1A and PIK3CA genes were only identified in oCCC patients, which is in line with previous reports (5, 23, 52). We also found one TP53 variant in an oCCC patient, which is unusual for oCCC that normally exhibits TP53 wild-type (25). Our finding may be explained by the fact that the tumor sample from this specific patient displayed a mixed clear-cell and endometroid histology although with dominant oCCC histology. In the tissue sample analyzed, both histology types may be present, thereby explaining the observation of the TP53 variants, although uncommon in clear cell, being reported in endometroid OC (25). Furthermore, all nine TP53 variants were previously reported in the IARC database and seven of them were present in the TCGA dataset (c.97-1G>A, c.413C>T, c.646G>A, c.659A>C, c.701A>G, c.731G>A, c.772G>A). In addition to TP53, we identified 33 variants, of which 5 were classified as “P” and 28 as “VUS”. In our opinion, it is relevant to report not only pathogenic variants with a documented role in the disease, but also VUSs, at least those that some pathogenic potential can be indicated, e.g., based on pathogenicity prediction bioinformatic tools. There is still a large number of variants revealed by NGS with undefined clinical significance, which accounts for about 40% of total variants (53). As variant classification may alter over time, periodical checks of the VUS outcomes might be necessary.
Our study has a few limitations. We decided to employ tumor-only testing based on our current settings to identify somatic variants, which is based on filtering variants against available databases (30). Therefore, our findings might be further validated by matched tumor and blood/adjacent normal tissue parallel testing, where somatic tumor-only variants are identified by subtraction of blood/normal tissue variants from the total variants found in the tumor tissue (30).
In conclusion, by using a discovery cohort of relatively few patients but investigating a total of 356 DDR genes using exome sequencing with high coverage, we have identified 42 somatic mutations that were not only limited to TP53, BRCA1/2, and HR-associated genes. They may display a potential role as biomarkers to predict platinum-based chemotherapy or PARPi treatment response, disease severity (measured on OS) or progression, as differences in disrupted DDR pathways between HGSC_LongOS, HGSC_ShortOS, and oCCC groups were observed.
Acknowledgements
The Authors would like to thank Mikael Kronborg Christophersen for performing whole exome sequencing experiments. The Authors thank the Bio- and Genome Bank Denmark for providing samples used in this study. The Authors received financial support from: The Mermaid Foundation, available at: http://www.mermaidprojektet.dk/ (JLJ, DNPO, CKH and EVH), Danish Cancer Research Foundation, available at: http://www.dansk-kraeftforsknings-fond.dk/ (EVH), and Herlev Hospital Research Council, available at: https://www.herlevhospital.dk/forskning/ (EVH).
Footnotes
Authors’ Contributions
All Authors participated in data analysis, discussed the results, and contributed to the writing of the final manuscript.
Conflicts of Interest
The Authors declare that there are no conflicts of interest in relation to this study.
- Received February 17, 2023.
- Revision received March 3, 2023.
- Accepted March 6, 2023.
- Copyright © 2023 International Institute of Anticancer Research (Dr. George J. Delinasios), All rights reserved.
This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY-NC-ND) 4.0 international license (https://creativecommons.org/licenses/by-nc-nd/4.0).





{kind=link}
{kind=link}