


Abstract
Background/Aim: Glioblastoma (GBM) is the most common and most lethal type of cancer of the central nervous system in adults. Despite aggressive treatment, which is based on surgical resection, if possible, followed by radiation and chemotherapy, a high recurrence rate and therapy resistance is observed. Thus, additional innovative therapies are urgently needed to improve the poor median survival of only 15 months. Treatment of solid tumours with non-invasive physical plasma (NIPP) represents such a novel and innovative anticancer procedure. Materials and Methods: In this study, we investigated the effect of NIPP, an ionized argon gas, on the in vitro growth of human GBM cell lines, LN-18 and U-87 MG. Proliferation was measured by live cell count. Subsequently, proliferative factors were analysed at the level of nucleic acids (polymerase chain reaction) and proteins (western blotting). Results: For both GBM lines, a treatment time-dependent decrease in growth was observed compared to controls. Additionally, NIPP treatment resulted in reduced rates of AKT serine/threonine kinase 1 (AKT1) and extracellular-regulated kinase 1/2 ERK1/2 expression, whereas expression of p21, proliferating cell nuclear antigen, and heat-shock proteins 90α and 90β was not affected. In both cell lines, a strong increase in expression of tumour-suppressive microRNA-1 (miR-1) was detected after exposure to NIPP. Conclusion: Our results demonstrated that NIPP is able to efficiently attenuate growth of GBM cells and suggest AKT1, ERK1/2 and miR-1 to be pivotal factors of NIPP-modulated cellular signalling. Translated into the clinical setting, NIPP may represent a promising option for the treatment of GBM.
Glioblastoma (GBM) represents the most aggressive brain tumour of glial origin and accounts for approximately 60% of all brain-derived tumours. The invasive and nearly always lethal phenotype of GBM is particularly characterized by behaviour of rapid and infiltrative growth deep into the healthy brain tissue (1-3). Despite multimodal therapy consisting of surgical resection followed by chemotherapy and radiation, the median survival is still only about 15 months. In spite of the investigation of new GBM treatment approaches, no substantial improvement in GBM therapy has been achieved in recent years (4, 5).
A promising and novel therapy option is the treatment of tumours with non-invasive physical plasma (NIPP), previously also known as cold atmospheric (pressure) plasma, which primarily consists of an ionized gas. Unlike classical thermal plasma, the fourth state of matter, NIPP is not in a thermal equilibrium, the ions in the gas cloud have a much lower kinetic energy than the ions in classical thermal plasma. This makes it possible to generate the plasma at room temperature (6). NIPP is generated by high-frequency, high-voltage discharges between two electrodes. The high energy input ionizes the gas passing the electrodes and NIPP is formed (6). Currently, there are two main techniques used to generate NIPP. In the plasma jet principle, a gas, usually a noble gas, is ionized between two electrodes and emerges as NIPP effluent. In dielectric barrier discharge, on the other hand, the ambient atmosphere between the electrode and the grounded surface to be treated is used to form a NIPP (6-8).
According to the current understanding, the effect of NIPP is based primarily on the formation of reactive oxygen species (ROS) which interact with cellular structures (9). ROS are formed at the interface of plasma and ambient atmosphere. In tissue, ROS lead to the induction of oxidative stress in the extracellular matrix, as well as in intracellular compartments. Due to the application of NIPP, modifications of the extracellular matrix, the cell membrane, and intracellular components and structures are observed. In particular, changes in cytoplasmic membrane permeability and induction of DNA double-strand breaks have been detected (9, 10). Thereby, dose-dependent effects of NIPP have been described for some time. At low doses, proliferative and pro-angiogenic actions of NIPP have been demonstrated (10). At high doses, cell damage resulting in apoptosis and necrosis was found. In this regard, not only were antimicrobial effects detected, but also a strong selectivity of NIPP in impairing the growth of tumour cells was shown (6, 8, 9). A large number of potential applications of NIPP are currently in clinical and pre-clinical investigation. At present, most advanced is the research regarding the application of NIPP for the treatment of acute and chronic wounds. Other possible areas of application which are currently under research include dentistry, ophthalmology, and the large field of oncology (8, 9, 11).
Although some studies have investigated the effects of NIPP on GBM, the underlying molecular mechanisms are still poorly understood. We therefore evaluated the effect of NIPP treatment on an in vitro GBM model, consisting of the well-established cell lines LN-18 and U-87 MG. Molecular analyses included of growth and resistance-associated proteins serine/threonine kinase 1 (AKT1), p44/p42 mitogen-activated protein kinase (ERK1/2), cyclin-dependent kinase inhibitor 1 (p21), proliferating cell nuclear antigen (PCNA), heat-shock protein 90 (HSP90) and microRNA-1 (miR-1).
Materials and Methods
Cell culture and NIPP treatment. The maintenance of LN-18 and U-87 MG cells (both human GBM cell lines authenticated by the American Type Culture Collection, Manassas, VA, USA) was performed in Dulbecco’s modified Eagle’s medium supplemented with 10% foetal bovine serum, 2 mM glutamine and 1% non essential amino acid solution (100×) (all from PAN Laboratories, Cölbe, Germany) at 37°C with 95% humidity and 5% CO2.
For NIPP treatment, we used a Kinpen 09, a plasma jet manufactured by Neoplas tools (Greifswald, Germany). This physical plasma jet is operated with argon as carrier gas at a flow rate of 3 l/min. It creates a 9-13 mm long plasma effluent with a temperature below 42°C, and the sinusoidal operating frequency is approximately 1 MHz (12).
For cell growth experiments, 15,000 cells/well were seeded into a 24-well cell culture plate. In the experimental group, treatment with NIPP was performed for 5 s or 15 s. The control group was treated with non-ionized argon for the same time period. The treatment of cells with the Kinpen 09 jet was performed manually. The jet was placed at a distance of 9-13 mm above the cell suspension such that the tip of the effluent only reached the surface of the cell culture medium. Subsequently, the cells were transferred into new 24-well plates in order to exclude a possible influence of NIPP on the surface of the primarily used cell culture plate. Cell growth was determined after 4, 24, 48, 72, 96, and 120 h of incubation using a CASY TT cell counter and analyser (Roche Applied Science, Mannheim, Germany) with a 150 μm capillary, after detaching the cells with trypsin. The measurement was performed three times with 400 μl each in triplicate.
Western blotting. For analysis of the expression and phospho-activation of selected proliferation-related proteins (AKT1, ERK1/2, HSP90α, HSP90β, p21, and PCNA), the treatment time with NIPP was set to 15 s. Measurement of target proteins was performed after 24 and 48 h of incubation. For preparation of protein lysates, harvested cells were incubated on ice for 30 min with lysis buffer (50 mM Tris-HCl pH 7.4, 100 mM NaCl, 0.1% Triton X-100, 5 mM EDTA, 1 mM leupeptin, 1 mM aprotinin and 250 μg/ml sodium vanadate). BCA Protein Assay Kit (Thermo Fisher Scientific, Rockford, IL, USA) was used to determine protein concentration of cell lysates. After protein denaturation in sodium dodecyl sulphate (SDS) sample buffer (50 mM Tris-HCl pH 6.8, 2% SDS, 10% glycerol, 0.01% bromophenol blue, 5% 2-mercaptoethanol) at 95°C for 5 min, 30 μg of each protein sample were separated on an SDS polyacrylamide gel. For immunoblotting a tank blot system (Bio-Rad, Hempstead, UK) was used to transfer separated proteins onto a nitrocellulose membrane (GE Healthcare Life Science, Solingen, Germany). After blotting, membranes were blocked in 5% fetal bovine serum in Tris-buffered saline containing 0.05% Tween 20 (TBST) and 1% bovine serum albumin for 1 h at room temperature. For immunodetection, membranes were incubated with the primary antibodies (Table I) overnight at 4°C followed by a washing procedure with TBST. Incubation with the secondary antibodies was performed for one hour at room temperature again followed by washing step with TBST. For detection of chemiluminescent signals of horseradish peroxidase-coupled secondary antibodies, ChemiDoc XRS Imaging System (Bio-Rad) using ECL Plus Western Blotting Substrate (Thermo Fisher Scientific, Waltham, MA, USA) followed by densitometric analysis (Quantity One; Bio-Rad) was used. Fluorescence signals of IRDye-conjugated secondary antibodies were detected using an Odyssey CLx Imaging System (Li-cor Bioscience, Bad Homburg, Germany). The relative chemiluminescence or fluorescence intensities of the specific bands were calculated and normalized to those of glyceraldehyde 3-phosphate dehydrogenase as a loading control.
Antibodies for western blot analysis.
Quantitative reverse transcription-polymerase chain reaction (RT-PCR). RT-PCR was used for quantification of miR-1 expression after treatment of GBM cells with NIPP for 15 s, followed by a cell culture period of 24 and 48 h. For RNA isolation by phase segregation, peqGOLD TriFast (Peqlab, Erlangen, Germany) was used according to the manufacturer’s protocol. Total RNA was determined by NanoDrop 2000 spectrometer (Peqlab), and samples were adjusted to a content of 100 ng RNA/μl. Synthesis of cDNA coding for miR-1 and the housekeeping gene U6 was achieved by a Thermal Cycler T3000 (Biometra, Göttingen, Germany) performing stem-loop RT-PCR with primers targeting specifically miR-1 and U6 (miR-1 SLOOP, U6 SLOOP, Table II).
Oligonucleotides for quantitative reverse transcription-polymerase chain reaction analysis.
Nucleic acid quantification was performed by real-time PCR with a CFX96 Real-Time PCR Detection System (Bio-Rad). Amplification of miR-1 and U6-coding cDNA was achieved using specific forward- and reverse primers. Simultaneous fluorescence-based quantification was performed via SYBR-Green detection using SensiMix SYBR Hi-ROX Kit (Bioline, Luckenwalde, Germany) between the amplification cycles. The analysis of the quantitative PCR data was performed in CFX Manager 2.0 (Bio-Rad).
Statistics. Statistical analysis and graphical representation were performed using GraphPad Prism software 5.0 (GraphPad Software, Inc., San Diego CA, USA). Analysis of variance served as the test procedure for cell kinetics, and the Wilcoxon signed-rank test was used for analyses of western blot and RT-PCR data. A value of p<0.05 was considered to be statistically significant. Grubbs’ test for identification of outliers was performed using GraphPad QQuickCalcs Outlier calculator.
Results
Dose- and time-dependent growth-inhibitory effect of NIPP on GBM cells. In our study, we first evaluated the effect of NIPP on GBM cell proliferation by performing growth kinetics based on the measurement of living cells at 4, 24, 48, 72, 96 and 120 h after NIPP treatment. Both NIPP treatment durations led to a significant reduction in growth rate compared to the control group in U-87 MG cells (Figure 1A), as well as in LN-18 cells (Figure 1B). This growth-inhibitory effect was most pronounced after 72, 96 and 120 h. In addition, there was a clear increase in growth inhibition when the duration of NIPP treatment was increased from 5 to 15 s. This observation was consistent and similar in both GBM cell lines. The LN-18 growth kinetics showed 48% less cell growth after 5 s of treatment, while after 15 s of treatment, a decrease of 87% was observed. The results for U-87 MG matched these findings, with growth rate reductions of 44% and 69%, respectively.

Comparison of the effects of non-invasive physical plasma (NIPP) on cell growth of U-87 MG (A, B) and LN-18 (C, D) cells. Cells were treated for 5 (A, C) or 15 s (B, D) with NIPP or the carrier gas (argon; control). Living cells were assessed applying a CASY Cell Counter and Analyzer (OLS, Bremen, Germany) at 4, 24, 48, 72, 96, and 120 h after treatment. Data are given as mean and standard deviation (U-87 MG n=6, LN-18 n=5), two-way ANOVA with Bonferroni post-hoc test. Significantly different at: p<0.05; **p≤0.01; ***p≤0.001.
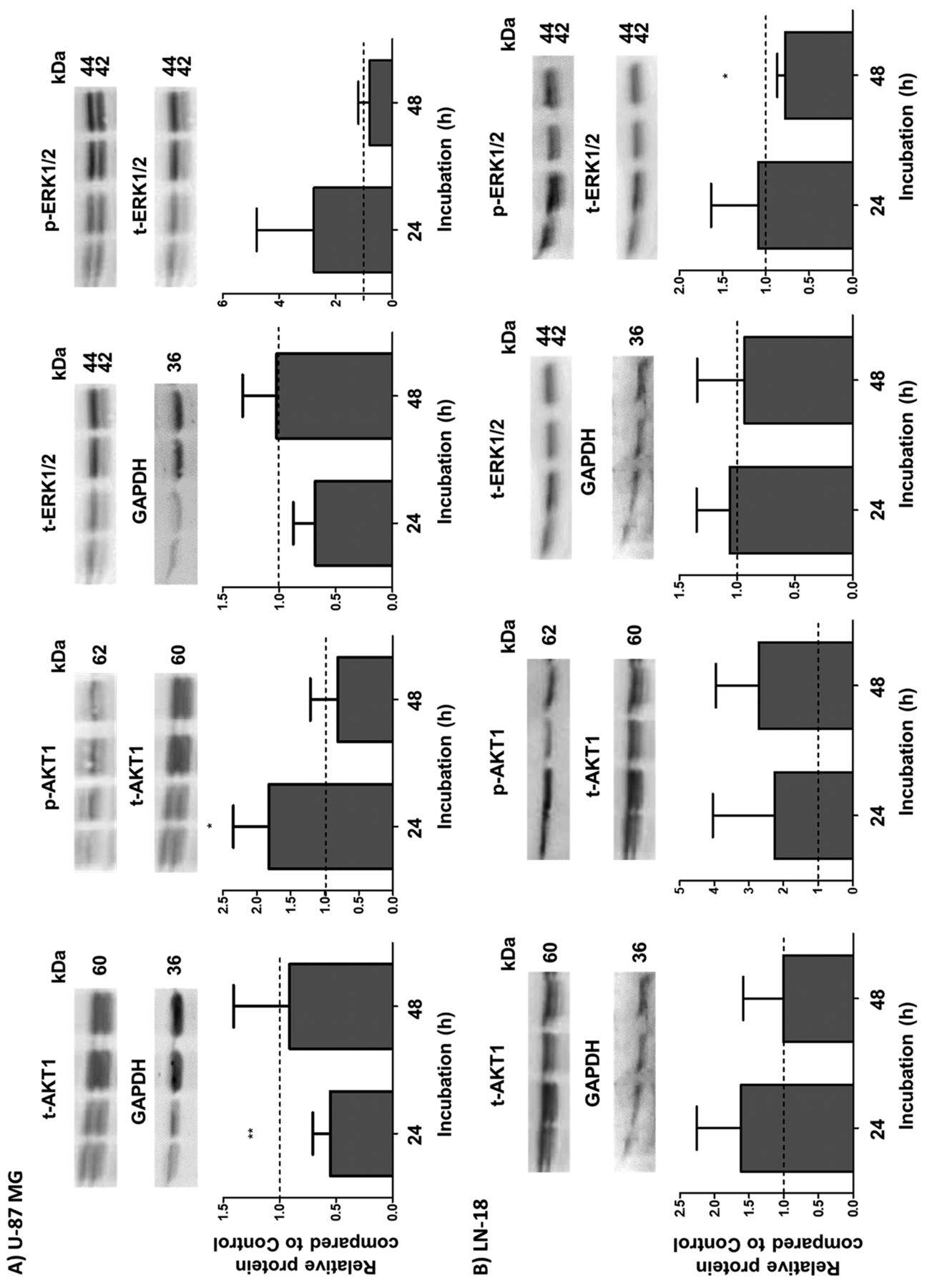
Reduction of the AKT1 and ERK1/2 protein expression after NIPP treatment. To evaluate which signalling pathways might be affected by NIPP treatment, we analysed the expression and phospho-activation of two proliferation-related kinases: AKT1 and ERK1/2 (13, 14). Figure 2 represents the expression and activation data of both AKT1 and ERK1/2 in U-87 MG (Figure 2A) and LN-18 (Figure 2B) cells.

Western blot analyses of total (t-) and phosphorylated (p-) forms of serine/threonine kinase 1 (AKT1) and extracellular-regulated kinase 1/2 (ERK1/2) in U-87 MG (A) and LN-18 (B) glioblastoma cells. Cells were treated for 15 s with non-invasive physical plasma (NIPP) or carrier gas (argon; control) and protein expression was determined 24 and 48 h after NIPP treatment. Expression of t-AKT1 and t-ERK1/2 was normalized to the level of the housekeeping protein glyceraldehyde 3-phosphate dehydrogenase (GAPDH). p-AKT1 and p-ERK1/2 were normalized to the content of t-AKT1 and t-ERK1/2, respectively. The upper panels show representative blots in each case; the lower panels present the statistical analyses of 4-8 independent experiments as the mean (relative to that of control cells) and standard deviation. Significantly different at: *p<0.05 and **p≤0.01 by Wilcoxon signed-rank test.
U-87 MG cells showed a significant decrease (by half) in the expression of total AKT1 24 h after treatment with NIPP (Figure 2A). At the same time, the amount of phosphorylated, active AKT1 (p-AKT) was significantly increased (1.83-fold) by NIPP treatment (Figure 2B). However, 48 h after NIPP treatment, the relative levels of both activated and total AKT1 had returned to normal in U-87 MG cells. A similar pattern was observed for total ERK1/2 and its phosphorylated form (p-ERK1/2), with reduction of total ERK expression to 0.68 and a 2.77-fold increase in active p-ERK1/2 24 h after NIPP treatment of U-87 MG cells (Figure 2A), but this was not statistically significant (p=0.0625).
In LN-18 cells, on the other hand, there was a slight (1.5- to 2.0-fold) but not significant increase in both the total and active AKT1 level after NIPP treatment. The total EKR1/2 expression was not significantly changed by NIPP but p-ERK1/2 was slightly reduced to 0.78 48 h after NIPP treatment of LN-18 cells (Figure 2B).
Expression of HSP90α, HSP90β, p21, and PCNA is not affected after NIPP treatment. Since it is well-known that HSP90 promotes GBM cell viability by interaction with a wide range of client proteins to influence growth, survival, and the cell cycle (15), we next analysed HSP90α and HSP90β expression at 24 and 48 h after NIPP treatment. As shown in Figure 3, in both U-87 MG (Figure 3A) and LN-18 (Figure 3B) cells, no significant change in either HSP90α or HSP90β expression was detectable. Furthermore, protein expression of p21 and PCNA was also not significantly changed, despite a trend for a reduced PCNA content in U-87 MG cells 48 h after NIPP treatment.
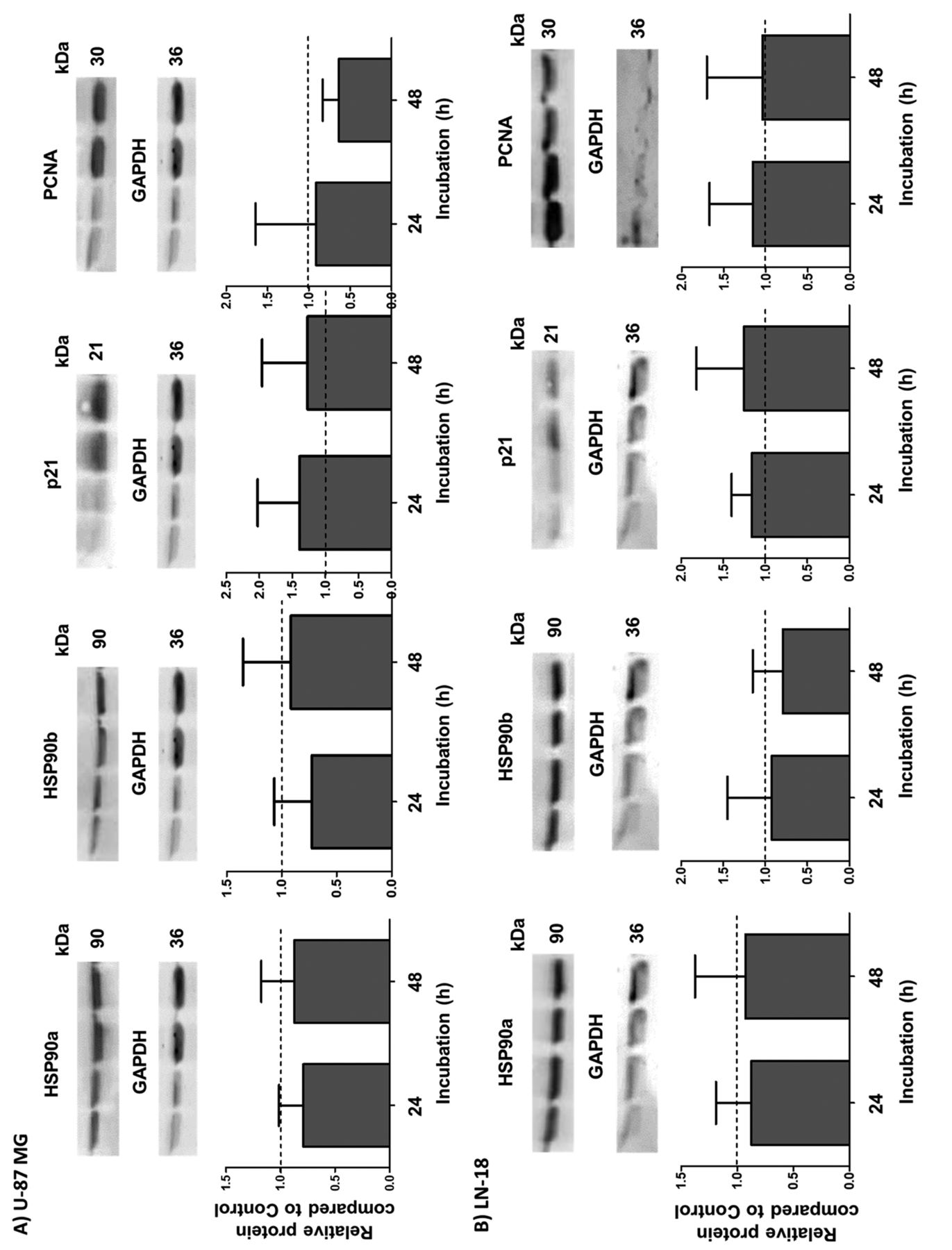
Western blot analyses of heat-shock protein 90α (HSP90α), HSP90β, p21, and proliferating cell nuclear antigen (PCNA) in U-87 MG (A) and LN-18 (B) glioblastoma cells. Cells were treated for 15 s with non-invasive physical plasma (NIPP) or carrier gas (argon; control) and protein expression was determined 24 and 48 h after NIPP treatment. Expression of proteins was normalized to the level of the housekeeping protein glyceraldehyde 3-phosphate dehydrogenase (GAPDH). The upper panels show representative blots in each case; the lower panels present the statistical analyses of 4-8 independent experiments as the mean (relative to that of control cells) and standard deviation. There were no significant differences by Wilcoxon signed-rank test.
Expression of tumour-suppressive miR-1 is elevated after NIPP treatment. An additional regulator of tumour progression is the tumour suppressor miR-1. As shown in Figure 4, 24 h after NIPP treatment a significant, almost 15-fold increase of the miR-1 level in U-87 MG cells and a more than 5-fold up-regulation in LN-18 cells was observed. Interestingly, 48 h after NIPP treatment, a decrease of the miR-1 content to the level of the controls was detectable in both GBM cell lines (Figure 4).
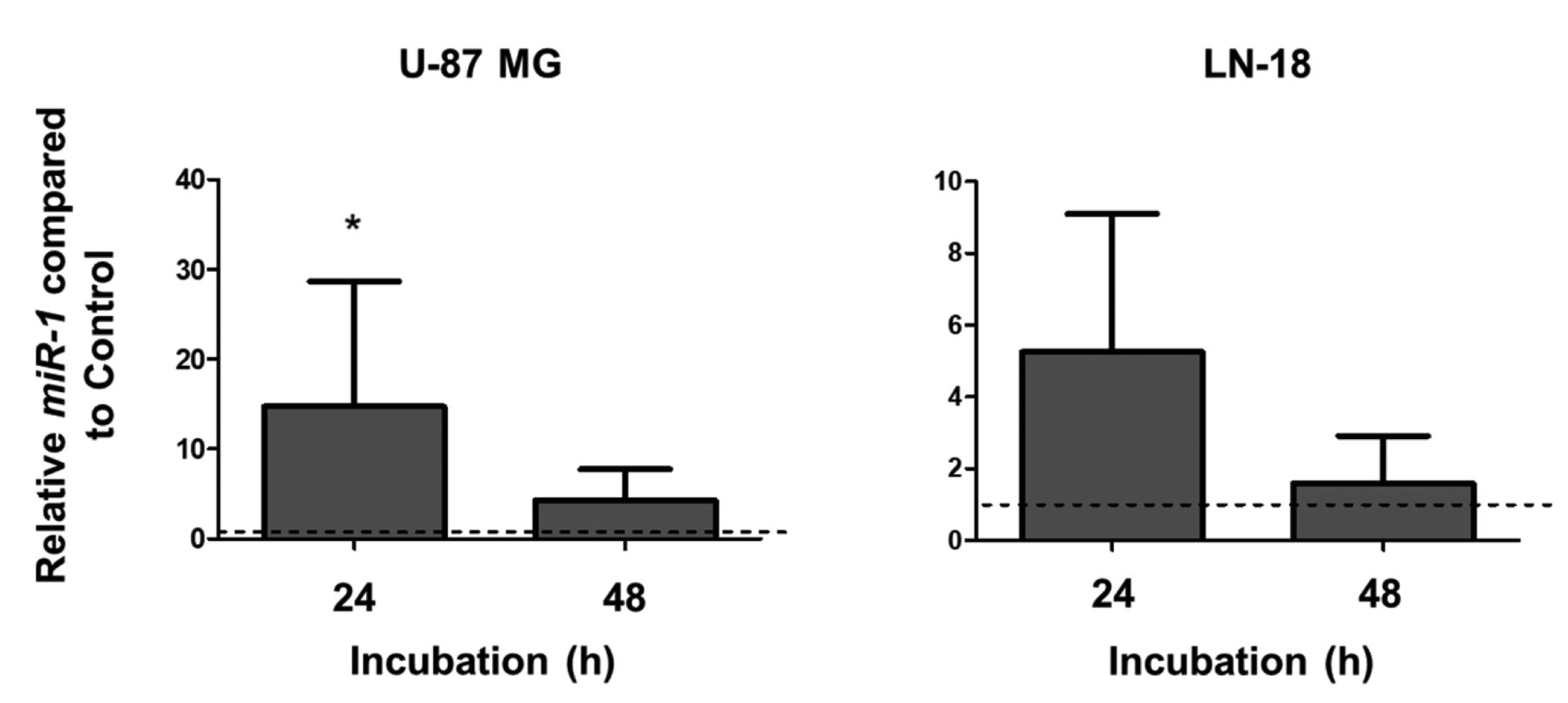
Expression of miR-1 in U-87 MG (left) and LN-18 (right) glioblastoma cells. Cells were treated for 15 s with non-invasive physical plasma (NIPP) or carrier gas (argon; control) and miR-1 expression was determined 24 and 48 h after NIPP treatment using quantitative reverse transcription-polymerase chain reaction analysis. miR-1 expression was normalized to that of endogenously expressed U6 snRNA. Data are given as the mean and standard deviation (n=5-7). *Significantly different at p<0.05 by Wilcoxon signed-rank test.
Discussion
GBM is one of the most deadly and difficult-to-treat brain cancers because of its infiltrative growth behaviour. Complete tumour removal by surgery is nearly impossible. Due to therapy resistance, patients with GBM experience relapse within a few months, resulting in a short median overall survival of only about 15 months. This emphasizes the urgent need for novel GBM therapies. NIPP has emerged as potential and innovative anti-tumoural option for solid tumours.
NIPP has been investigated for its suitability in several applications, including the treatment of cancer (16, 17). The treatment of cancer has diversified with the use of NIPP, which was shown to induce apoptosis in many types of cancers, including melanoma, pancreatic, ovarian, breast cancer or lung cancer (18-22). Some studies evaluating the tumour-suppressive effect of NIPP on GBM cells also exist (23-26), however, the influence of NIPP on the regulation of intracellular signalling cascades and growth-regulatory proteins is poorly understood. In in vitro experiments, an increased ROS content, intracellularly and extracellularly, as well as a restoration of sensitivity of O6-methyl-guanine DNA methyltransferase-positive tumour cells to temozolomide was demonstrated (6, 27, 28). More importantly, the first in vivo study also showed a significant reduction in growth of GBM in mice when treated with NIPP (26).
In this study, we investigated the influence of NIPP treatment on growth of two human GBM cell lines, LN-18 and U-87 MG. Additionally, we analysed the expression of key intracellular factors (AKT1, ERK1/2, HSP90α, HSP90β, p21, PCNA and miR-1) which are known to be relevant for GBM growth and progression.
In our study, we demonstrated a significant growth-inhibitory effect of NIPP on GBM cell models. Such an anti-tumoural impact of NIPP was already described in the literature using different GBM cell lines and affirms the potential application of NIPP as a therapeutic option for treatment of patients with GBM (23-25, 28, 29). As partly shown by others, we also confirm the dose- and time-dependent effects of NIPP treatment. The earliest significant reductions in cell number were found 72 h after NIPP application to LN-18 and U-87 MG cells, an effect which was much stronger after 96 and 120 h of incubation. Additionally, treatment of GBM cells for 15 s was more effective than that for 5 s. The strong growth-inhibitory effects of NIPP treatment were associated with a regulation of signalling molecules relevant for tumour growth and progression which will be discussed in detail below.
NIPP-induced growth suppression was accompanied by a significant change in the regulation of the pro-oncogenic survival kinase AKT1, which is an effector kinase of the phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K)-AKT pathway. Strong evidence has revealed that AKT1 also plays a major role in GBM progression and aggressiveness (30). AKT1 is a key protein associated with anti-apoptotic processes, proliferation, and migration (31). In many tumour types, including GBM, AKT1 expression has been found to be elevated during progression. Thus, AKT1 is discussed as a crucial factor in tumourigenesis (13). We found a decrease in total AKT1 expression in U-87 MG cells 24 h after NIPP treatment. Due to its important role in cell survival, this reduction of AKT1 expression may be directly caused by NIPP-mediated cytotoxic effects. But unexpectedly, the amount of phosphorylated, active AKT1 was increased 24 h after NIPP treatment. Whether this was actually an increase in the activity of AKT1 or only a relative excess due to the reduced total AKT1 content cannot be answered by our experiments. Furthermore, 48 h after NIPP treatment, the levels of both total and p-AKT1 returned to those of the control. This may mean that NIPP causes a short-term change in regulation of AKT1 which might be sufficient to induce other growth-regulatory cascades. The temporary increase in active AKT1 protein might also represent a rapidly activated survival cascade to protect GBM cells from death, which then cannot be maintained due to further cell damage processes caused by NIPP. Interestingly, in LN-18 cells, no significant change in expression of AKT1 by NIPP was found in our study, but there was a trend for increased total and active AKT1. Discrepancies between LN-18 and U-87 MG cells might be based on the different status of phosphatase and tensin homologue (PTEN), which is negatively regulates the activation of AKT1 (32). While LN-18 express wild-type PTEN, U-87 MG cells are described as PTEN-negative, which might be the reason for the observed significant increase in active AKT1 in U-87 MG caused by the lack of PTEN-mediated inactivation of AKT1 (33). At later time points, it is conceivable that other factors control AKT1 activation such as integrin-linked kinase, which has been shown to regulate AKT1 activity in PTEN-mutant GBM cells (34, 35). Regulation of AKT1 by NIPP is not described in the literature, but oxidative stress, which is thought to be induced by NIPP, was shown to inactivate PI3K/AKT1 signalling, while activity of its negative regulator PTEN appears not to be regulated directly (36, 37).
Besides AKT1, a role of ERK1/2 in GBM pathogenesis is discussed (38). A trend for a similar pattern of regulation was found for ERK1/2, which also belongs to the survival kinases, in U-87 MG cells. Again, there was a slight down-regulation of total ERK1/2 protein but an enhanced level of ERK1/2 phosphorylation 24 h after NIPP treatment. An increase in ERK1/2 phospho-activation has been already shown after treatment of melanoma cells and myeloblasts with NIPP (39, 40). In contrast, reduced total and active ERK1/2 were found in NIPP-treated prostate cancer cells (41). Thus, modulation of ERK1/2 by NIPP seems to be cell type-dependent. As a component of the RAS-ERK signalling cascade, ERK1/2 is an oncogenic kinase capable of influencing a variety of effectors, which are particularly associated with anti-apoptotic processes, cell growth and tumour progression (42, 43). Thus, an increased short-term activation of ERK1/2 by NIPP may represent a mode of protection for GBM cells to escape cell death as described for AKT1 above, which, however, cannot be maintained by additional cellular stress processes. Again, the observed changes of ERK1/2 in U-87 MG cells were not found in LN-18 cells. In contrast, there was a slight, but significant reduction in the level of p-ERK1/2 in LN-18 cells at 48 h after NIPP treatment. Once more, the different PTEN status of U-87 MG (PTEN-mutant) and LN-18 (PTEN wild-type) might be responsible for the different response of these two GBM cells to NIPP. Intact PTEN, described for LN-18 cells, is able to reduce ERK1/2 activation through inhibition of RAS, which can counteract the activation of ERK1/2 in LN-18 cells (44). In PTEN-mutant U-87 MG cells, there is no inhibition of RAS and thus also not of ERK1/2, which may conceivably lead to a greater increase in ERK1/2 activity.
Both the PI3K/AKT1 and mitogen-activated protein kinase/ERK1/2 pathways, as prominent drivers of GBM cell proliferation, are strongly dependent on HSP90 protein (15). Under chemotherapy, HSP90 promotes GBM cell viability and stabilizes inhibitors of apoptotic mechanisms. Thus, HSP90 contributes to chemoresistance (45, 46). Generally, HSP90 is considered a molecular chaperone responsible for correct folding, stabilization, and transport of proteins (47, 48). Two HSP90 isoforms are mainly expressed: the stress-induced HSP90α and the constitutively expressed HSP90β type (47). In many tumours, including GBM, expression of HSP90 was observed to be elevated (45, 49). Numerous studies have demonstrated that inhibition of HSP90 reduces the growth of GBM cells (50, 51). However, tumour-selective cleavage of HSP90 by oxidative stress and NIPP have also been reported (52, 53), but no data exist on GBM regarding regulation of HSP90 by NIPP or oxidative stress as far as we are aware. In our study, no significant changes of HSP90 isoforms α and β were detected in LN-18 and U-87 MG cells after NIPP application. Thus, HSP90 appears not to be a target of NIPP-mediated growth-inhibitory effects in GBM cells.
Both apoptosis and cell-cycle arrest are found to be induced by NIPP in tumour cells (54, 55). Expression analyses of the apoptosis-related and cell cycle-modulating proteins p21 and PCNA, which are also significantly involved in DNA replication and DNA repair, also showed no significant changes after NIPP application in both LN-18 and U-87 MG cells (56, 57). Due to the growth-inhibitory effects of NIPP, a change in expression of the cell-cycle regulator p21 after NIPP treatment would have been conceivable. In contrast to our results, melanoma, colorectal and prostate cancer cells treated with NIPP demonstrated a significant modulation of p21 (41, 58). This argues for a cell type-dependent regulation of p21 expression after NIPP exposure and may link to p21-independent growth inhibition in GBM cells. Regarding PCNA, a cell type-specific regulation was found after NIPP treatment. Whereas an increase of PCNA expression was described in human gingival and osteoblast-like cells (59, 60), modulation of PCNA was lacking in squamous cell carcinoma and malignant melanoma cells (61). In line with these findings, analysis of PCNA expression in our study also revealed a lack of change in PCNA expression after NIPP treatment of GBM cells. PCNA is responsible for a fast and smooth process of DNA synthesis and the initiation of DNA repair processes (59). There is currently much discussion whether NIPP-induced ROS directly cause DNA damage or only induce apoptosis or other forms of cell death (62).
miRs play pivotal roles in tumourigenesis, apoptosis, and drug resistance in cancer cells due to their regulatory function in post-transcriptional gene expression (62). Among them, the tumour-suppressive miR-1 has been shown to be predominantly down-regulated in almost all human cancer types, including GBM (62, 63). miR-1 suppresses the expression of factors that promote cell growth and angiogenesis and suppress apoptosis (64). To the best of our knowledge, this is the first study to demonstrate a strong increase of miR-1 after NIPP application in both LN-18 and U-87 MG cells. Thus, miR-1 up-regulation may represent a molecular mediator responsible for the tumour-suppressive effects of NIPP. Remarkably, the increase in miR-1 expression was only temporary and expression returned to the control level after 48 h of incubation, however, this short-term induction in miR-1 expression might be sufficient for the subsequent signal cascades to inhibit cell growth. Moreover, miR-1 has been classified as having a high potential to inhibit tumour progression and therapy resistance (65). Recently, Bronisz and colleagues showed that ectopic expression of miR-1 in GBM cells inhibited cell growth, neovascularisation, and invasiveness in vivo (63). miR-1 seems to have an important role as an effector molecule for inhibition of the aggressive growth behaviour of tumour cells such as GBM.
Conclusion
NIPP represents a new and promising treatment option for therapy of a variety of tumours, including GBM. Although most studies are still in the experimental stage, there are initial studies that demonstrate benefits of its application in patients. Nevertheless, there still seems to be a long way to go before clinical NIPP application can find its way into routine oncological therapy. Of note, a challenging part of treating GBM is that it is present in the brain, and any damage to the tissue during surgery or other treatment can cause a series of devastating effects in the central nervous system. Thus, the non-invasive set-up of NIPP, as suggested by Soni and colleagues (67), would be an important improvement for therapeutic options.
To date, many of the intracellular processes induced by NIPP in tumour cells have not been adequately studied or understood. Our study argues for a possible role of kinases, and especially of miR-1, as potential effectors of the growth-inhibitory effect of NIPP. This should be the subject of further research to improve the extremely poor survival of patients with GBM.
Acknowledgements
The Authors thank Lena Zeyda for her dedicated experimental support and Christian Scharf for his interested guidance of the NIPP experiments.
Footnotes
Authors’ Contributions
Conceptualization, S.B.M., M.B.S., S.L.; methodology, S.L., S.M., S.B.; formal analysis, S.L., S.B.M., S.B. and M.B.S.; investigation, S.L., S.B.M., S.M., S.B. and M.B.S.; writing - original draft preparation, S.L., S.B.M., H.W.S.S., A.M. and M.B.S.; writing - review and editing, S.L., S.B.M., H.W.S.S., A.M. and M.B.S.; visualization, S.L., S.B.M. and M.B.S.; supervision S.B.M. and M.B.S.
Conflicts of Interest
The Authors declare that they have no competing interests.
- Received October 8, 2022.
- Revision received November 10, 2022.
- Accepted November 14, 2022.
- Copyright © 2023 International Institute of Anticancer Research (Dr. George J. Delinasios), All rights reserved.
This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY-NC-ND) 4.0 international license (https://creativecommons.org/licenses/by-nc-nd/4.0).





{kind=link}
{kind=link}
{kind=link}
{kind=link}