


Abstract
Medulloblastoma (MB) is the most common malignant pediatric posterior fossa tumor. Recent genetic, epigenetic, and transcriptomic analyses have classified MB into three subgroups, Wingless Type (WNT), Sonic Hedgehog (SHH), and non-WNT/non-SHH (originally termed Group 3 and Group 4), with discrete patient profiles and prognoses. WNT is the least common subgroup with the best prognosis, characterized by nuclear β-catenin expression, mutations in Catenin beta-1 (CTNNB1), and chromosome 6 monosomy. SHH tumors contain mutations and alterations in GLI1, GLI2, SUFU, and PTCH1 genes, which constitutively activate the SHH pathway. Originally, the presence of TP53 gene alterations and/or MYC amplifications was considered the most reliable prognostic factor. However, recent molecular analyses have subdivided SHH MB into several subtypes with distinct characteristics such as age, TP53 mutation, MYC amplification, presence of metastases, TERT promoter alterations, PTEN loss, and other chromosomal alterations as well as SHH pathway-related gene mutations. The third non-WNT/non-SHH MB (Group3/4) subgroup is genetically highly heterogeneous and displays several molecular patterns, including MYC and OTX2 amplification, GFI1B activation, KBTBD4 mutation, GFI1 rearrangement, PRDM6 enhancer hijacking, KDM6A mutation, LCA histology, chromosome 10 loss, isochromosome 17q, SNCAIP duplication, and CDK6 amplification. However, based on molecular profiling and methylation patterns, additional non-WNT/non-SHH MB subtypes have been described. Recent WHO (2021) guidelines stratified MB into four molecular subgroups with four and eight further subgroups for SHH and non-WNT/non-SHH MB, respectively. In this review, we discuss advancements in genetics, epigenetics, and transcriptomics for better characterization, prognostication, and treatment of MB using precision medicine.
Medulloblastoma (MB) is a malignant embryonal tumor that is composed of small, round blue cells arising from the cerebellum or brainstem and exhibiting a high mitotic index and nuclear pleomorphism. MB originates from lower rhombic lip progenitor cells during infancy or early childhood, perhaps due to impaired differentiation (1, 2).
MB is the most common malignant pediatric primary brain tumor accounting for approximately 20% of all childhood brain tumors and 63% of intracranial embryonal tumors (3). These tumors can manifest in infants, children, and young adults, with an overall annual incidence of approximately 5 cases per 1 million in the pediatric population.
MB most commonly, about 70%, occur in children under 10 years of age. MB is approximately 1.7 times more common in males than females, however, sex predilection as well as age-specific incidence differ by subgroup (2). MB is rare in adults, with a reported incidence of 0.05 cases only per 100,000 population (4). All MBs are classified as WHO Grade IV. Originally classified based on histological features, MBs are now classified primarily by molecular features in addition to histologic features (5). There are four main molecular subgroups of MB: Wingless Type (WNT), Sonic Hedgehog (SHH), non-WNT/non-SHH Group 3 (Group 3), and non-WNT/non-SHH Group 4 (Group 4). Group 3 and Group 4 (Grp3/4) are now considered as part of a larger heterogenous subgroup termed non-WNT/non-SHH. WNT and SHH subgroups are associated with activation of the corresponding molecular signaling pathways. The heterogeneous Grp3/4 subgroups have their own discrete genetic and epigenetic characteristics. Each of the four MB subgroups are associated with different patient profiles, prognoses, and molecular features. Since the WHO consensus (2016), multiple investigations have studied the genetic, epigenetic, and transcriptomic profiles of MB tumors and identified further subtypes of the four primary subgroups. Based on the genetic and epigenetic analyses, a variety of subdivisions have increased understanding of MB behavior. The subtypes have different survival, age, sex, rates of metastasis, histology, and molecular features. DNA methylation profiling is now generally considered the gold-standard for identifying MB subtypes and for evaluating the heterogeneity within these subgroups, however, a new international consensus is yet to be established.
MB is initially diagnosed based on clinical symptoms as well as magnetic resonance imaging (MRI) of the brain with evidence of the primary tumor, CSF cytology to assess for microscopic metastases, and an integrated pathological and molecular analysis from surgically obtained tumor tissue. Current standard-of-care therapy for MB includes maximal safe surgical resection, cytotoxic chemotherapy, and radiation therapy. The 2016 consensus for subgroups, subtypes, or risk stratification of MB poorly handles the heterogeneity within the four primary MB subgroups as well as within those cohorts with standard or high-risk features. The disparities among patient outcomes within the same subgroups suggest the utility of further subdivisions or subtypes of MB, which are addressed in current 2021 guidelines (6).
WNT-activated Subgroup
The WNT group overall has the best prognosis out of all the MB groups (2). Several studies have presented evidence for at least two distinct risk stratifications or subtypes within the WNT subgroup, while others report only one. One potential subtype has a low-risk profile with >90% survival. This lower-risk WNT subtype is typically non-metastatic, affects those <16 years old, and commonly possesses a chromosome 6 monosomy. The other WNT subtype may include adult patients, metastatic cases, and large cell/anaplastic (LCA) histology (7-9). As different groups have used different criteria and techniques for subdivision of the WNT subgroup, each study reports different survival, metastatic percentage, and age data.
WNT MBs are associated with accumulation of nuclear beta-catenin, mutations in CTNNB1, which are found in 86% (60-90%) of WNT MB, and chromosome 6 monosomy that may be found in 83% (80-90%) of WNT MBs (10-14). CTNNB1 mutation, and monosomy 6 are frequently the only genetic changes tested for identifying WNT MB, therefore it is estimated that potentially 10-15% of WNT MB tumors could be missed (14). ALK alterations were found in 12.7% (7/55) of MBs, with at least 57% (4/7) of ALK positive tumors belonging to WNT MB (15). In addition, 100% (48/48) of tumors without ALK positivity in their study were classified as Grp3/4 (15).
Other recurrent mutations include DDX3X (36%), CSNK2B (14%), EPHA7 (8%), SMARCA4 (19%), ARID1A (8%), ARID2 (SWI/SNF nucleosome-remodeling complex) (8%), TP53 (14%), KMT2D (14%), PIK3CA (11%, BAI3 (8%), SYNCRIP (8%), and ATM (6%) (2). APC gene mutations are associated with Turcot syndrome and can be seen in about 8% of WNT subtype tumors (14). Mutations in APC binding sites AXIN1 and AXIN2/Conductin have also been reported (16).
Multiple methods have been described for subtyping WNT MBs. Some studies do not have any further subdivision of WNT (8). Clinical subdivisions based on the presence or absence of metastatic disease have been suggested for WNT MB tumors (2). Other groups have used both gene expression and DNA-methylation arrays and an analysis technique called similarity network fusion (SNF) to create two subdivisions of WNT MB. Cavalli et al. (7), applied SNF and spectral clustering and identified two subtypes of WNT MB, termed WNTα and WNTβ. WNTα affects mainly children and predominantly has monosomy 6, whereas WNTβ affects older patients and monosomy 6 is a rare event (6/21).
SHH-activated Subgroup
As the name suggests, the SHH MB is associated with genetic abnormalities of the Sonic Hedgehog pathway. SHH MB is derived from cerebellar granular neuronal precursors (GNPs), which in normal brain development migrate from the external germinal layer to the internal granule layer under the control of the SHH pathway (1, 2, 17). During normal development, the Purkinje cells of the cerebellum secrete Shh, leading to inhibition of Patched1 (Ptc1), releasing its brake on Smoothened (SMO), which allows for transcription of Gli proteins (18-20). Once these GNPs have migrated to their final position, SHH pathway activity stops (21). However, due to mutations in the components of this pathway, the development of SHH MB can occur.
SHH-activated MBs display a bimodal age distribution, occurring most commonly in infants and adults and to a lesser degree in children, with a 1.5:1 male-to-female ratio (22). SHH-activated MB originates most commonly in the lateral cerebellum and accounts for approximately 30% of all MBs (1). However, some studies (23) have also found that about 53% of SHH MBs were hemispheric, with the remaining SHH MBs involving the vermis. The location of MB can be age-dependent as it has been shown that those in older children and young adults were present predominantly in the rostral cerebellar hemispheres, whereas in infants they are more frequently observed in the vermis (24).
About half of SHH MB are nodular/desmoplastic types, which are rarely observed in other genetic subtypes; classic and LCA subtypes are also observed in SHH MBs (2). Early array-based expression studies identified active SHH signaling in a subset of MBs, which were often desmoplastic/nodular (D/N) or MB with extensive nodularity (MWEN). Specialized reticulin staining is commonly used to assess desmoplasia. Internodular desmoplasia was needed for a diagnosis of D/N/MB, including the paucinodular D/N variant, and MBEN. In addition, dispersed small nodules amid widespread desmoplasia was seen in the paucinodular D/N MB (11, 12). Intranodular cells in this variant especially demonstrate the differentiated neurocytic morphology of the conventional D/N tumor or MBEN. These cells express neuronal proteins and display low Ki-67 immunostaining. The characteristics of MBEN includes its large irregularly shaped nodules, pronounced internodular neurocytic differentiation, as well as sparse internodular desmoplastic regions (11, 12).
An extensive mRNA expression profiling analysis established the presence of the SHH group, and SHH is internationally accepted as one of the four main molecular groups of MBs (25). In addition, genome-wide methylation profiling, performed in formalin-fixed tissue, has further reinforced SHH as a distinct group (14). Furthermore, smaller sets of SHH-related genes determined by gene-counting technology or qRT-PCR have supported the use of this molecular group for the clinical classifications.
Previously, the WHO guidelines had reported a SHH subgroup with TP53 mutations and MYC amplifications as the main predictors of prognosis, but many other molecular features have been uncovered since then, leading to a better understanding of the heterogeneity within the SHH group (2, 7-9). Various investigators have attempted to further stratify MBs into additional subtypes. Cavalli et al. (7), categorized SHH into SHHα, SHHβ, SHHγ, and SHHδ subtypes based on methylation profiling. SHHβ and SHHγ affect predominantly infants, whereas SHHα and SHHδ mostly children and adults, respectively. SHHα and SHHβ have greater metastatic potential in comparison with SHHγ and SHHδ subtypes. Ramswamy et al. (26), reported three risk categories for SHH MB. Standard risk is considered wild-type TP53, no MYCN amplification, and non-metastatic. High-risk SHH MBs are metastatic, have MYCN amplification, or have both features. Very high-risk SHH MBs have TP53 mutations and may be either metastatic or non-metastatic. In contrast, Schwalbe et al. (8), reported only two cohorts of SHH MBs, with age as one of the main differences between the subtypes. The study only considered patients from infancy to 16 years of age and divided the group into two cohorts based on presence or absence of a metagene V4. The infant-SHH cohort was predominantly <4.3 years of age and the older cohort was predominantly ≥4.3 years. The infant-SHH cohort had a higher metastatic rate, but also had better survival than the child-SHH cohort. Both cohorts had SUFU and PTCH1 mutations, but the child-SHH cohort more commonly had TP53 mutation, TERT mutation, and MYC and GLI2 amplification. Yet another group divided SHH into further subtypes. Northcott et al. (2), defined two low-risk SHH subtypes (one SHH-MBEN and one iSHH-subtype II), two intermediate-risk SHH subtypes (one SHH/p53wt and the other SHH, non-MBEN), one high-risk SHH subtype (iSHH-subtype I), and one very high-risk SHH subtype (SHH/p53t). Furthermore, gain of chromosome 2 was found predominantly in the iSHH-I group, whereas PTCH1 deletions due to loss of chromosome 9q were enriched in the second group, iSHH-II (27). These results were in accordance with the findings of Robinson et al. (28) on these subtypes, showing similar chromosomal aberrations. In addition, MYCN amplification did not distinguish these subtypes, and only one patient with MYCN amplification was found in the iSHH-I subtype. Both iSHH-I and iSHH-II displayed equal distribution of DMB and MBEN histological subtypes. Other genetic alterations, such as mutations in PTCH1, SUFU, and SMO genes were equally distributed among both subtypes. Interestingly, neither subtype expressed TP53 gene mutations. Only one patient who had a PTCH and TP53 variant of uncertain significance has been reported (27). No significant major survival differences between patients with iSHH-I and iSHH-II have been detected.
There have been multiple mutations associated with SHH MBs, including PTCH1, suppressor of fused homologue (SUFU), Smoothened (SMO), and GLI (1, 2). The key molecular drivers of SHH pathway genes in MBs include mutations and other genetic alterations in known pathway genes in 116 (87%) of 133 SHH tumors (29). In addition, structural variations or mutations in genes are often distinct across the MB molecular groups. High-level amplifications associated with the SHH group include loci containing MYCL, GLI2, PPM1D, YAP1, and MDM4. MYCN expression is often amplified in SHH MBs, and can be directly upregulated by GLI transcription factors in both granule cell precursors and tumors. An important effector of the SHH signaling pathway is YAP1 in both neoplastic and non-neoplastic cerebellar progenitors. In addition, homozygous deletions at the PTEN and PTCH1 loci are observed only in SHH tumors, which are not seen in other molecular groups of MB. An investigation of coding regions in 92 primary MB and normal sample pairs, revealed that somatic mutations are relatively rare in MB as compared to other tumors with a median of 12 non-silent and 4 silent mutations (30). Similarly, SHH tumors are exclusively associated with mutations in PTCH1, SUFU, and other genes of the SHH pathway (31). Nevertheless, SHH pathway activation can be observed in some SHH tumors, without any obvious genetic basis. One of most frequent somatic point mutations are in the TERT promoter, causing an enhanced telomerase activity. An investigation of 466 MBs demonstrated TERT mutations in 21% of all tumors, with the highest frequency among adult cases of the SHH group (83%; 55 of 66 cases) and better outcomes (32).
An inherited syndrome associated with SHH MB is the Gorlin syndrome, also known as nevoid basal cell carcinoma syndrome. It is associated with multiple basal cell carcinoma (BCC) of the skin, odontogenic jaw keratocytes, MB, and other developmental abnormalities. The nevoid BCC syndrome is due to heterozygous germline mutations in PTCH1 as seen in 97 of 171 patients (56%), indicating that mutations in PTCH1 and TP53 are mutually exclusive. It is important to note that the age distributions of these groups are also distinct as MB with germline PTCH1 or SUFU mutations are observed in infants and children aged < 4 years, whereas those with a germline p53 mutation occur in older children (33). PTCH1 was mapped to chromosome 9q22 by the linkage analysis (18, 34, 35). Loss of heterozygosity in this region has also been demonstrated in many sporadic desmoplastic/nodular MB. PTCH1 is an inhibitor of SHH signaling and is especially vital in cerebellar development.
Patients with SHH/TP53wt MB are commonly children aged <4 years, adolescents, or young adults, while those with SHH/TP53mt MB are mostly children aged 4-17 years (29). An investigation of 133 SHH activated MBs revealed that 28 patients (21%) with a median age of 15 years had a TP53 mutation (24). Genetic syndromes with TP53 mutations have been found in a larger proportion in the SHH group MB.
SHH/TP53mt MB are commonly associated with large cell/anaplastic morphology and loss of chromosome 17p. These tumors also show an amplification of GLI2, MYCN, or SHH, whereas mutations in PTCH1, SUFU, and SMO are generally absent. SHH TP53mt tumors can be associated with germline or somatic mutations in the negative regulators PTCH1 or SUFU, as well as activating somatic mutations in SMO or (rarely) amplification of GLI2. In addition to genetic changes activating SHH signaling, mutations in DDX3X or KMT2D and amplification of MYCN or MYCL are sometimes observed, as are deletions of chromosomal arms 9q, 10q, and 14q (36).
Point mutations in TP53, especially in SHH and not in WNT MB are associated with chromothripsis, a process in which chromosomes shatter and acquire multiple rearrangements simultaneously in a single catastrophic event. Overall, the frequency of specific genetic changes in SHH MBs seems to be somewhat different in infants, children, and adults (37). A TP53 germline mutation is commonly detected in MB patients >3 years of age. BRCA2 and PALB2 gene abnormalities show no strict association with age at diagnosis. Germline point mutations in TP53, as seen in Li-Fraumeni syndrome, can result in SHH MBs. Patients with SHH/p53mt MB are considered a distinct high-risk group.
Mutations in TP53 are commonly found in the DNA binding regions of the gene, which is also known as the hotspot region of the gene that encodes exons 4 through 8. TP53 mutation can be used to segregate the different MB subtypes into favorable and high-risk subtypes. One other aspect of SHH/p53mt is the high rate of amplification of MYC. Approximately half of all SHH/p53mt MBs have been shown to have germline rather than somatic alterations (24, 38). Recent updated WHO classification has clearly defined SHH MB into two major categories: 1) SHH MB TP53wt and 2) SHH MB TP53mt (6).
Non-WNT/Non-SHH-activated Subgroup
WHO classification (2016) has combined Grp3/4 into one subgroup of MB termed non-WNT/non-SHH subgroup MBs, because these subgroups are not due to an anomaly of a single functional pathway (5), like WNT or SHH MB. Grp3/4 MBs share remarkable similarities in their genetic alterations (2). Similar to SHH, Grp3/4 MBs are genetically heterogeneous, although some tumors share common genetic abnormalities. Also, a mixture of Grp3/4 features have also been identified within the non-WNT/non-SHH MB (2, 7, 8, 13).
Classic histology type is mostly observed in non-WNT/non-SHH MB. Also, non-WNT/non-SHH MB tumors infrequently display areas of rosette formation or a palisading pattern of tumor cell nuclei. Similarly, these tumors can show nodule formation in the absence of desmoplasia. In addition, LCA characteristics of tumors in the non-WNT/non-SHH group mostly belong to Group 3. Moreover, the neural marker immunohistochemical (IHC) profile of non-WNT/non-SHH MB is similar to that of the WNT or SHH groups displaying classic or LCA tumors. The tumors express synaptophysin to a variable extent and are rarely immunopositive for GFAP. Non-WNT/non-SHH tumors show cytoplasmic but not nuclear β-catenin immunoreactivity. A diagnostic method involving a distinct set of antibodies (GAB1, β-catenin, filamin A, and YAP1) was able to distinguish WNT and SHH MBs from non-WNT/non-SHH MB subgroups (12). Another four-antibody approach used DKK1 for WNT, SFRP1 for SHH, NPR3 for Grp3, and KCNA1 for Group 4 MB, allocating 98% of samples into each subcategory; however, such diagnosis by IHC remains limited without genetic analysis.
Group 3 Medulloblastoma
Group 3 MB make up approximately 20% of all MB cases and about 45% of cases in infants (39). They are typically LCA, usually affect infants and children, and are more common in men. One unique feature of the presentation of Grp3 tumors is a high rate (40%) of metastatic disease at the time of diagnosis. Over-expression of MYC through amplification or aberrant expression is more common in Grp3 MB in comparison with other groups. Amplification of MYC has long been known as a genetic alteration associated with poor outcome in all patients with MB, but it has been reported to have independent prognostic significance in Grp3 tumors. MYC encodes the proto-oncogene C-MYC, which has an important role in cell cycle progression, apoptosis, and cellular transformation, as discussed earlier in reference to SHH subgroup MB. In addition, MYC-PVT1 fusion was associated with MYC over-expression (2).
There are several prevalent somatic alterations in Grp3 MB. KBTBD4 has several hotspot somatic mutations (6%); KBTBD3 encodes Kelch repeat and BTB domain containing 4, a protein of unknown function. Recurrent alterations in Grp3 include SMARCA4 (9%), CTDNEP1 (5%), KMT2D (5%), and GFI1 (4%). Growth factor independent 1B (GFI1B) is a transcription factor involved in the development and differentiation of hematopoietic cell lines. In approximately 11% of Grp3 MB there is a phenomenon known as GFI1B enhancer hijacking, whereby structural variants on chromosome 9q reposition highly active enhancer regions present in DDX31 near GFI1B, resulting in GFI1B over-expression. DDX31 encodes DEAD-box helicase 31, a protein of unknown function. OTX2 (7.7%) and LRP1B (4.6%) are also over-expressed in Grp3 MB (40, 41). There is a preponderance of CD133 expression (42), isochromosome 17q, 1q gain, 7 gain, 10q loss, and 16q loss in Grp3 MB (2, 22).
Some mutations are present in multiple subtypes of MB, especially in Grp3/4 MBs. Specifically, GFI1 and GFI1B are inserted next to active enhancers in Grp3/4 MB, leading to their activation, known as “enhancer hijacking” (43). In addition, some Grp3/4 MBs have mutations in KDM6A histone modifier, deregulation of H3K27me3 chromatin repression, EDH2 over-expression, CHD7, and gain of chromosome 7q (31). In addition, CTDNEP1, also known as DULLARD, is mutated in some Grp3/4 MBs and is associated with 17p deletion (2). One of the most common chromosomal changes in Grp3/4 MB involve copy number alterations on Chromosome17: 17p deletions, 17q gain, or a combination of these in the form of an isodicentric 17q. The methylome profiles of Grp3 MBs are more similar to each other than to WNT or SHH group MBs (2). Grp3 MBs are known to have poor prognosis (22).
Group 4 Medulloblastoma
Group 4 MBs, the largest molecular subgroup, make up about 40% of all MB tumors (2). They are associated with an intermediate prognosis like SHH MB and affect all age groups (peak incidence seen at 5-15 years) (22). Grp4 MBs are much more common in men. While they tend to be of the classic histologic subtype, they can also be LCA. One third of Grp4 MB patients have metastasis at time of presentation (2), which is associated with poor outcome and is one of the most robust prognostic markers among Grp4 tumors. There are several prevalent somatic alterations in Grp4 MB. PRDM6 encodes PR/SET domain 6 protein, which contains a transcriptional repressor with potential histone methyltransferase activity. Many Grp4 MBs have PRDM6 enhancer hijacking (17%), due to recurrent structural variants on chromosome 5q that reposition enhancers in the SNCAIP (synuclein-alpha interacting protein) region close to PRDM6, resulting in increased PRDM6 expression. MYCN amplification (6%), encoding the neuroblastoma MYC oncogene transcription factor, is another common feature of Grp4 MB. There are also hotspot somatic mutations in KBTBD4 (6%), and recurrent alterations in the following genes: KDM6A (7%), OTX2 (6%), ZMYM3 (6%), KMT2C (6%), ZIC1 (4%), and CDK6 (4%). Group 4 tumors (~80%), like group 3 tumors, also have chromosome 17 copy number alterations but in a much larger proportion. There may be also loss of X chromosome or mutations in the X chromosome genes ZMYM3, CHD7, KDM6A, and KDM1A (2, 31). ZMYM3, CHD7, and KDM6A are chromatin remodeling genes; for example, KDM6A is responsible for regulating methylation of H3K27 (44). A subset of non-WNT/non-SHH tumors that do not cluster reliably into Grp3/4 show activated GFI oncogenes.
Molecular Methods for Classification of Non-WNT/non-SHH MB
Due to common genetic abnormalities in Grp3/4, 2016 WHO guidelines have combined these subtypes into a new subgroup termed as non-WNT/non-SHH MB (5). Epigenetics, primarily methylation patterns, have been used by several groups to subtype non-WNT/non-SHH MB in several different ways. As in the previous consensus findings for Grp3/4 tumors, several molecular patterns have been found to be associated with the various subtypes of non-WNT/non-SHH MB, including but not limited to MYC amplification, MYCN amplification, GFI1B activation, KBTBD4 mutation, GFI1 rearrangement, OTX2 amplification, PRDM6 enhancer hijacking, KDM6A mutation, LCA histology, chromosome 10 loss, isochromosome 17q, SNCAIP duplication, and CDK6 amplification.
Some groups attempted to risk stratify non-WNT/non-SHH or Grp3/4 tumors based on a combination of findings. For example, Ramaswamy et al. (26) noted at least two risk stratifications of Group 3 MBs and at least three stratifications of Group 4 MBs. Group 3 consisted of a standard risk cohort (no metastasis and no MYC amplification) and a very high-risk cohort (metastatic). Other Group 3 MBs might have MYC amplification without metastasis, isochromosome 17q, or anaplasia on histology. Group 4 was divided into a low-risk cohort (chromosome 11 loss with no metastasis), a standard risk cohort (without chromosome 11 loss and with no metastasis), and a high-risk cohort (with metastasis).
Schwalbe et al. (8) (Total n=428; Grp3/4 n=267) subtyped all MBs into seven molecular subtypes using consensus-NMF with resampling by DNA-methylation array, to analyze methylation patterns. WNT was considered a subgroup without further subtyping. SHH MB was split into two age-related subtypes. Based on the High risk (HR) and Low risk (LR) Grp3/4 MBs, each split into a high and low risk category, with clear evidence for shared biology. Grp 3 and Grp 4 were divided into two subtypes each, and designated as Grp3-HR, Grp3-LR, Grp4-HR, and Grp4-LR.
Cavalli et al. (7) (n=763; Grp3/4; n=356), using SNF analysis for both gene expression and DNA-methylation profiles, classified Grp3/4 MB into three subtypes (a, b, and γ) for each Grp 3 and Grp4, making it total 6 subtypes of Grp3/4/. They identified twelve total MB subtypes: WNT was split into two subtypes (α and β), SHH into four subtypes (a, b, γ, and d), and Grp3/4 into six subtypes, each split into three subtypes (a, b, and γ). Northcott et al. (14) (Total n=1256; n=740 Gr3/4) used tSNE/DBSCAN methodology to analyze methylation patterns and subdivided Grp3/4 MBs into eight non-WNT/non-SHH subtypes. These techniques separate data into natural clusters and generate a distinctive profile of each cluster established by the most important characteristics. Importantly, in line with the previously described evidence of shared behavior, at least three of these eight subtypes exhibited shared Grp3/4 behavior. With such diversity of classification, the results of these several analyses of Grp3/4MBs were combined using the following methods. Northcott et al. (14) used t-SNE/DBSCAN techniques to assess the DNA-methylation array in a large series of 1,256 MBs (14). This approach was a combination of the following techniques: t-Distributed Stochastic Neighbor Embedding (t-SNE), a dimension reduction technique, and density-based clustering of applications with noise (DBSCAN). These approaches complemented each other as t-SNE supported and preserved local variations between samples and global disparities between the subgroups, and the DBSCAN rendered the clustering algorithm based on proximity (or density), which led to the identification of eight molecular subtypes of Grp3/4 (n=740). The use of different methodologies produces different numbers of subtypes each with different propensities for the different common Grp3/4 molecular alterations. Sharma et al. (45) (n=1501 Grp3/4 MBs) took the molecular data for the patients from the three studies described above and resampled and analyzed the data to classify non-WNT/non-SHH tumors into eight subtypes (Table I).
Demographic, histological, and genetic characteristics of non-WNT/non-SHH MB subtype subtypes (I-VIII).
Metastatic disease in MB portends a poor prognosis. MB is staged according to the Chang classification, which defines macroscopic and microscopic metastases, ranging from M0 to M4. M0 tumors have no evidence of metastasis on imaging or CSF studies. M1 tumors have no evidence of metastasis on imaging, but CSF studies detect tumor cells. M2 tumors have intracranial metastatic deposits beyond the primary site. M3 tumors have metastatic deposits in the spinal subarachnoid space. M4 metastatic MB tumors are characterized by having metastasis outside the cerebrospinal axis. Clinical management of these tumors includes risk-adapted craniospinal irradiation (CSI) and adjuvant chemotherapy after the maximal neurosurgical resection. Such approach has produced the best survival outcome for MB patients, with an estimated 5-year overall survival of ~80% for non-infants with non-metastatic GTR, and ~60% for children with metastatic MB and/or STR (Figure 1).
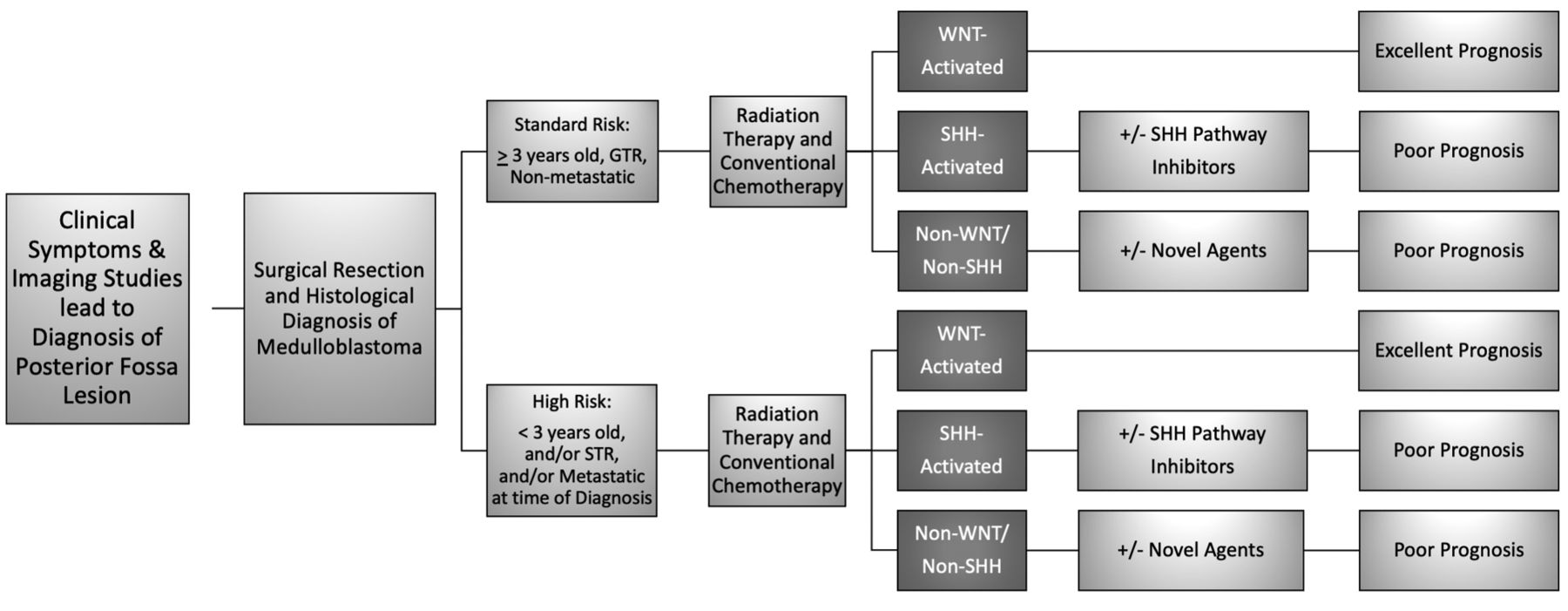
Clinical risk stratification, prognoses, and management strategies. Diagram depicting standard of care management and expected prognosis [adapted from Sharma et al. (45)]. Standard-of-care therapy for medulloblastoma (MB) includes surgical resection followed by cytotoxic chemotherapy, and cranio-spinal irradiation (CSI) in patients ≥3 years of age. Clinical outcome of disease strongly depends on patient’s age, histopathological features of tumor as well as the genetic profile. Standard-risk patients are >3 years of age, with gross total resection (GTR), and non-metastatic disease (70-85% overall survival for 5 years). High-risk patients are generally <3 years of age, have subtotal resections (STR), and/or metastatic disease at the time of diagnosis (<70% overall survival for 5 years).
Each MB subgroup exhibits distinct prognoses and responses to chemotherapy and radiation. WNT subgroup of MB comprises approximately 10% of all MB cases and they have the best overall prognosis with >95% survival at 5 years. In WNT MB, several recurrently altered genes have been identified, including DDX3X (in 36% of patients), SMARCA4 (19%), TP53 (14%), and PIK3CA (11%). Several of these genetic alterations can be found in a wide variety of human cancers, and the clinical applications of finding these genetic alterations are a matter of ongoing study and the targets of clinical trials. Several clinical trials (NCT02066220, NCT01878617, NCT02212574 and NCT02724579), which study de-escalation of therapy for low or standard risk WNT MB, are ongoing. Children >3 years of age treated with conventional multi-modal therapy and children with WNT MB <16 years of age at time of diagnosis have a favorable prognosis when treated using current standard of therapy. Careful reduction in dosage is currently being tested in clinical trials, however, such a protocol is yet to be defined for patients older than 16 years.
Several clinical trials have used molecular subgroups and clinical risk categories for treatment selection and investigated therapy de-escalation for low-risk WNT MB (NCT01878617). Also, a phase II trial investigated the effectiveness of maintenance chemotherapy with reduced radiation therapy on average-risk WNT MB, as characterized by positive nuclear beta-catenin expression using, CTNNB1 mutations, and absence of MYC and MYCN amplifications (NCT02724579). In addition, the SIOP PNET 5 phase II/III trial (NCT02066220) evaluated the efficacy of risk-dependent treatment adjustments, with the aim to confirm high event-free survival in patients with a low-risk profile defined by non-metastatic MB with total and near-total tumor resections, lack of LCA histology, absence of nuclear beta-catenin immune positivity, and absence of MYC and MYCN amplifications. In addition, other treatments for low-risk WNT MB patients include radiotherapy with a dose of 54 Gy to the primary tumor and 18.0 Gy to the craniospinal axis without chemotherapy, which is generally followed by 6-cycles of reduced-intensity maintenance chemotherapy.
Approximately 30% of all MBs are SHH MB and fall into an intermediate prognosis category. SHH MB patients harbor either germline or somatic mutations, or copy-number alterations in critical genes associated with SHH signaling pathway. These genes include PTCH1 (43%), SUFU (10%), SMO (9%), GLI1 or GLI2 amplifications (9%), and MYCN amplifications (7%). Most adult SHH MB patients (80%) harbor PTCH1 or SMO mutations. In addition, alterations in components of the TP53, such as focal copy-number variations, are observed in SHH MB patients. In addition, activation of receptor tyrosine kinase (RTK)–PI3K pathways has also been reported in SHH MB. Moreover, mutations in TERT promoter region have been detected in ~39% of patients. Prognosis for SHH MB is correlated with age, metastatic staging, and the presence of certain molecular features, such as TP53 gene alterations, MYC amplification, and aberrant PI3K/mTOR pathway signaling (46). In general, deregulation of TP53 signaling disrupts cell cycle regulation, apoptosis, and DNA repair pathways, whereas aberrant PI3K signaling promotes cellular growth, proliferation, and survival. For SHH MB, it is recommended to perform at least a minimal mutational analysis of PTCH1, SUFU, and TP53 in tumor and blood. In older adolescents with SHH MB, there is a prevalence of germline and somatic TP53 mutations. The presence of germline and somatic TP53 mutations is infrequently evident in infants and adults. Amplification of MYCN and/or GLI2 are often coincident with TP53 loss-of-function mutations and/or deletions. In contrast to patients with WNT MB with TP53 mutations who have an excellent prognosis, those with SHH MB and germline or somatic TP53 mutations have poor survival. Infants with tumors of desmoplastic medulloblastoma (DMB) and medulloblastoma with extensive nodularity (MBEN) histology typically is SHH/TP53wt and have favorable results, in spite of the fact that the treatment included chemotherapy only protocols that spare CSI. MYC amplification and presence of metastatic disease are uniformly regarded as high-risk features. Additional preclinical studies have shown that high-risk MYC-driven MB can be treated with a combination of HDAC and PI3K inhibitors. Other preclinical data suggest that GFI1- and/or GFI1B-driven MB can be treated with the combination of cell cycle checkpoint inhibitors and cytotoxic chemotherapy, or CDK inhibitors. In addition, therapeutic targeting of lysine-specific demethylase 1 (LSD1) with or without cytotoxic agents have also been considered.
LSD1 specifically demethylates mono- or di-methylated histone H3 lysine 4 (H3K4) and H3 lysine 9 (H3K9) via a redox process and has been shown to be elevated in some subsets of MB (47). It has been shown that LSD1 plays an important role in cellular processes, including cell proliferation, adipogenesis, spermatogenesis, chromosome segregation, and embryonic development, and may be involved in tumor progression, in part by inhibiting the tumor suppressor activity of TP53.
Therapeutic Strategies for MB
Therapeutic strategies can be adapted for subtypes of MB patients based on their discrete genetic abnormalities. Numerous dysregulated signaling pathways are implicated in the pathogenesis of MB. Currently, targeting multiple signaling pathways is being considered in the treatment of MB, some of which are depicted in Figure 2.
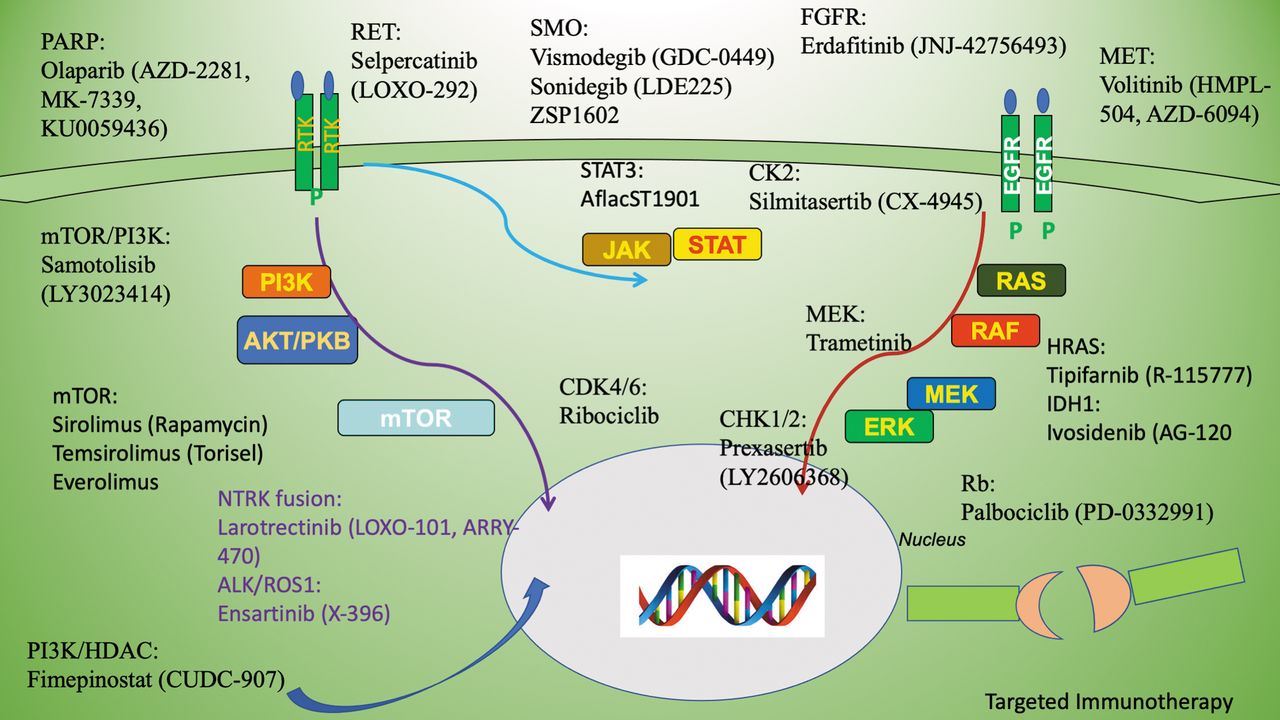
A schematic depiction of multiple signaling pathways representing discrete RTKs and other growth factor signaling effectors. PI3K: Phosphatidylinositol 3’-kinase; mTOR: mechanistic target of rapamycin; EGFR, Epidermal growth factor receptor; RET: Rearranged during transfection; SMO: smoothened; FGFR: fibroblast growth factor receptor; MET: MET proto-oncogene; HDAC: histone deacetylases; STAT3: signal transducer and activator of transcription 3; MEK: mitogen-activated protein kinase kinase; HRAS: Harvey Rat sarcoma; CHK1/2: checkpoint kinase ½: CDK4/6: cyclin-dependent kinase 4/6): Rb: retinoblastoma proteins; PARP: Poly (ADP-ribose) polymerase.
SHH MB adult patients tend to have a higher burden of genome wide single nucleotide variant (SNV)s compared to younger SHH MB patients. Approximately >80% of adult SHH MB patients harbor alterations in either PTCH1 or SMO. Considering that these mutations occur upstream in the SHH pathway, adults SHH MB patients are excellent candidates for targeted therapeutics based on molecular profiling, such as inhibitors of SMO that have shown promising results in clinical trials. In fact, one ongoing study currently stratifies patients in molecular subgroups (NCT01878617) and adds an SMO inhibitor to the standard treatment for SHH MB patients. Additionally, Vismodegib (GDC-0449), a small molecule inhibitor of SMO, has demonstrated efficacy against refractory MB in preclinical studies (48). However, resistance to targeted Vismodegib therapy is observed in infants and children with frequent mutations downstream of SMO, especially those affecting SUFU or GLI1. Inhibition of SMO can hamper normal development of bone and tooth in pediatric patients that can lead to a growth plate fusion which can stay long even after the end of therapy.
Recently Menyhart and Gyorffy treated MB based on molecular alterations in discrete subtypes (48). Preclinical strategies for targeted therapies against SHH MB are mainly geared towards epigenetic treatments with BET bromodomain inhibitors, G2/M regulators (AURK and PLK), and cMET inhibitors (48). In addition, strategies are also developed to target stem-like cells, as well as further interventions using immunomodulatory therapies. The feasibility and toxicity of oral Vismodegib maintenance therapy following conventional chemotherapy was explored for standard or high risk newly diagnosed MB in skeletally mature SHH-activated patients in a phase II trial (NCT01878617). In addition, other treatments and drug combinations are available for recurrent/refractory SHH-activated MB. Also, a combination therapy using the CDK4/6 inhibitor Ribociclib and SMO inhibitor Sonidegib (LDE225) for adult SHH-activated patients, who had specific gametic alterations such as 9q copy number loss or PTCH1 mutations, was examined in a phase I trial by the St. Jude Children’s Research Hospital (NCT03434262). WNT- and SHH-activated MBs are being treated with Ribociclib and the MEK-inhibitor Trametinib. Furthermore, the effect of CX-4945, silmitasertib sodium, a small molecule inhibitor of Casein kinase II (CK2), has been evaluated in a Pediatric Brain Tumor Consortium Phase I/II trial (NCT03904862) in skeletally immature (phase I) or mature (phase II) SHH-activated recurrent/refractory MB patients. In this investigation, the subgroup determination was confirmed by a CLIA certified methylation-based assay. In addition, a targeted therapy using the CHK1/2 inhibitor Prexasertib (LY2606368) in combination with cyclophosphamide for recurrent/refractory SHH-activated or Grp3/4 MB was examined in the St. Jude ELIOT trial (NCT04023669). Another trial (NCT03734913) explores the efficacy of ZSP1602, a SMO protein inhibitor, in the treatment of adult MB patients with advanced disease, irrespective of their SMO or Gli1 alteration status or molecular subgroup (49). Several other inhibitors have also been tested in clinical trials for the treatment of MB These include AflactST1901 (WP1066) for inhibition of STAT3 (NCT04334863), Selpercatinib (LOXO-292) for inhibition of RET (CT04320888), Tipifarnib (R-115777) for inhibition of HRAS (NCT04284774), Ivosidenib (AG-120) for inhibition of IDH1 (NCT04195555), Palbociclib (PD-0332991) for inhibition of Rb (NCT03526250), Erdafitinib (JNJ-42756493) for inhibition of FGFR (NCT03210714), Larotrectinib (LOXO-101, ARRY-470) for inhibition of NTRK fusion protein gene (NCT03213704), Ensartinib (X-396) for inhibition of ALK/ROS1 (NCT03213652), Olaparib (AZD-2281, MK-7339, KU0059436) for inhibition of PARP (NCT03233204), and Volitinib (HMPL-504, AZD-6094) for inhibition of MET (NCT03598244).
PI3K/AKT/mTOR Signaling Pathway and Associated Treatments
Somatic mutations in PIK3CA, an oncogene promoting constitutive activation of downstream AKT and mTOR signaling and resulting in aberrant cellular growth, proliferation, survival, as well as migration, are found in a wide variety of human cancers (48). Recurrent chromosomal alterations of the PI3K/AKT/mTOR pathway, including mutations in PIK3CA (7%), amplification of PIK3C2B (2%) and PIK3C2G (1%), and mutations in PTEN occur mainly in adult patients (50).
The PI3K/AKT/mTOR pathway has been shown to be deregulated in many cancers, including MB (46, 50). The mechanistic Target of Rapamycin (mTOR) is a serine/threonine protein kinase that frequently undergoes aberrant activation in cancer (50). mTOR forms two functionally distinct complexes (mTORC1 and mTORC2), to regulate various physiological functions. Inhibition of these complexes has been observed in MB (50, 51). Pharmacological inhibition of PI3K/AKT/mTOR pathway activity in combination with cisplatin provided potentially a novel opportunity to treat recurrent, chemoresistant MB patients as these therapies can lead to stimulation of FBW7 expression, degradation of SOX9 protein, and apoptosis (52). In fact, the combination of compounds targeting different cognate molecules in the PI3K/AKT/mTOR pathway leads to synergistic activity, as shown in preclinical studies (53). In addition, the PI3K/mTOR pathway is known to intercept with several cell signaling pathways allowing cancer cells to escape the inhibitory effect of PI3K/mTOR inhibitors. Consequently, inhibition of the PI3K pathway alongside other targets has been considered in the treatment of MB. In fact, a synergistic response of histone deacetylase inhibitor (HDACIs) with PI3K inhibitor has been shown in MYC-driven MB cells (53, 54). Based on these findings, new compounds to target different components of the PI3K/AKT/mTOR pathway simultaneously are in constant development (49) (Table II).
Clinical trials for MB with PI3K/Akt/mTOR signaling inhibition.
As described in Table II and Figure 2, several mTOR inhibitors are used in clinical trials for the treatment of recurrent/refractory MB with or without other signaling pathway inhibitors. Rapamycin analogs, such as Sirolimus, Temsirolimus, and Everolimus, are being generally used in phase I or phase II clinical trials. Samotolisib, a dual inhibitor of PI3K/mTOR, is currently in a phase II trial for the treatment of recurrent/refractory MB. In addition, a recently described dual inhibitor, Fimepinostat, which inhibits PI3K as well as histone deacetylase (HDAC) is in a phase I trial for the treatment of recurrent MB.
Targeted therapy trials for Grp3/4 MB are not as extensive as those targeting the WNT-activated and SHH-activated groups given the increased complexity of their genetic alterations. Group 3 non-WNT/non-SHH MB constitute 25% of MBs and have the poorest prognosis of all subgroups, due to a high proportion of tumors with MYC amplification. These tumors exhibit a high level of LCA histology and are the most likely subgroup to present with metastatic dissemination. Group 4 tumors are the most common subtype, accounting for 35% of MBs, and exhibit an intermediate prognosis similar to that of SHH MB. Grp3/4 MB can be reliably distinguished using DNA methylation- or expression-based approaches. The presence or absence of MYC or MYCN amplification may assist with risk stratification of Group 3 MB. Patients with Group 3 MB, especially those with MYC-amplified tumors and/or LCA histology, also have a dismal outcome as shown by multiple prospective clinical trials, whereas patients with Group 4 MB tend to have a more favorable prognosis. For example, Group 4 low-risk patients, or patients with either chromosome 11 loss or chromosome 17 gain have a very good prognosis. LCA variants comprise ~10% of cases and are associated with poor outcomes.
For infants with Group 3 MB, who have a very poor prognosis, there is an urgent need to integrate novel treatment options. More advanced treatment strategies to treat MB include chimeric antigen receptor (CAR) T-cell, vaccination, lineage targeting, or combinations of these (55). Current available therapies deserve future attention to provide advanced treatment of MB. In particular, immunotherapy advances are currently under investigation in many cancers including MB. In fact, potential therapeutic targets such as PRAME207 and HER2 have been investigated in several studies, as a novel means to target MB (56, 57). Furthermore, a clinical trial investigating the effects of HER2-specific CART cell therapy in patients with MB is currently ongoing (58). The treatment of this heterogeneous subgroup with tailored therapy along with reduced long-term toxicity remains a further challenge in the treatment of MB patients.
Certain clinical trials have been designed based on specific molecular subtypes of MB, such as the NCT01878617 trial of Prexasertib (LY2606368), the clinical trial (NCT04023669) of a targeted CHK1/2 inhibitor administered in combination with cyclophosphamide for Group 3, Group 4, and SHH-activated MB or gemcitabine for Grp3/4 MB. Another clinical trial (NCT03434262) combines the cyclin-dependent kinase inhibitor Ribociclib and Gemcitabine for Grp3/4 MB. Since the PI3K pathway is also upregulated in some Group 3 MB (53), down-regulation of this pathway might improve MB outcome. In fact, preclinical studies have also used inhibitors of PI3K and mTOR signaling pathways to assess the synergistic effect of PI3K and HDAC inhibitors, especially in patients with Group 3 MB (54).
Conclusion
While the identification of the four genetic subtypes of MB has helped categorize patients and predict the behavior of their disease, there is still much to be learned. Molecular subtyping has rendered further definition of subgroups of MB with certain defined genetic alterations that can be used for prognosis as well as diagnosis of disease to improve the management of MB. We have achieved a great deal of understanding in the genetic and epigenetic subclassifications of MB. However, there is still much to be accomplished in terms of transferring data from bench to bedside in the treatment of MB. With the development of drugs targeting the aberrant pathway of each MB subtype, there is hope that prognosis can improve for all patients with MB.
Acknowledgements
Supported by research funds from Brain and Spine Surgeons of New York and Advanced Research Foundation.
Footnotes
Authors’ Contributions
Writing – Original Draft: TS, JSR; Editing-Reviewing: AD; Resources: AM, MT; Supervision: CG, MJU; Concept development & Writing: MJU.
Conflicts of Interest
All Authors declare that there are no conflicts of interest in relation to this study.
- Received December 30, 2021.
- Revision received March 7, 2022.
- Accepted March 18, 2022.
- Copyright © 2022 International Institute of Anticancer Research (Dr. George J. Delinasios), All rights reserved.
This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY-NC-ND) 4.0 international license (https://creativecommons.org/licenses/by-nc-nd/4.0).
References
In this issue





{kind=link}
{kind=link}
Jump to section
- Article
- Abstract
- WNT-activated Subgroup
- SHH-activated Subgroup
- Non-WNT/Non-SHH-activated Subgroup
- Group 3 Medulloblastoma
- Group 4 Medulloblastoma
- Molecular Methods for Classification of Non-WNT/non-SHH MB
- Therapeutic Strategies for MB
- PI3K/AKT/mTOR Signaling Pathway and Associated Treatments
- Conclusion
- Acknowledgements
- Footnotes
- References
- Figures & Data
- Info & Metrics