


Abstract
Background/Aim: Olaparib was previously shown to synergistically enhance the cytotoxicity of DNA synthesis inhibitors in oesophageal carcinoma (OC) cell lines. However, the mechanisms of this synergy are not fully understood. As P53 binding protein 1 (53BP1) expression was previously shown to potentiate the anticancer effect of olaparib, we investigated the involvement of 53BP1 in the synergetic cytotoxic effects of olaparib and anticancer drugs in KYSE70 cells. Materials and Methods: Experiments included small interfering RNA transfection, growth inhibition assays, western blots, immunofluorescence, and flow cytometry. Results: The toxicity of DNA synthesis-inhibiting agents plus olaparib was decreased when 53BP1 was depleted. Olaparib cotreatment significantly increased phosphorylated H2A histone family member X (γH2AX) foci as well as 53BP1/γH2AX co-localisation in anticancer drug-treated cells. Silencing of 53BP1 suppressed anticancer drug-induced apoptosis with or without olaparib. Conclusion: Olaparib potentiates the cytotoxicity of anticancer drugs through 53BP1 in OC cells.
Oesophageal carcinoma (OC) is a malignant disease with a low 5-year survival rate (36%); moreover, 54% of patients have invasion and/or metastasis to other organs when diagnosed (1, 2). Therefore, cancer chemotherapy is essential to improve the outcomes of patients with this disease. Combination of 5-fluorouracil (5-FU) and cisplatin (CDDP) (FP therapy) and as well as FP plus docetaxel are typical first-line treatments that improve outcomes; the response rate to FP therapy is 35-40% in patients with OC (3, 4). However, 60% of patients fail to respond to treatment, and their options post first-line therapy are limited (5). Therefore, alternative anticancer chemotherapies are critical for improving the outcomes of patients with OC.
Poly (ADP-ribose) polymerase (PARP) inhibitors competitively inhibit PARP enzymatic activity and suppress DNA damage-repair and -response (DDR) pathways that are activated following single- or double-strand breaks (DSBs) (6, 7). The PARP inhibitor veliparib was found to prolong the median progression-free survival of patients with ovarian cancer when administered with paclitaxel and carboplatin therapy (8). Olaparib, the only PARP inhibitor clinically used in Japan, sensitizes OC cell lines to CDDP or radiation in vitro (9, 10). We previously discovered that olaparib synergistically enhanced the cytotoxicity of CDDP, doxorubicin, 7-ethyl-10-hydroxy-camptothecin (SN-38), and temozolomide via increased DNA damage in OC cell lines (11). Therefore, it is expected that PARP inhibitors may improve the outcomes of patients with this disease; however, the mechanisms by which PARP inhibitors elicit drug sensitization in OC cells are not fully understood.
A single administration of PARP inhibitor combined with cancer chemotherapy has been shown to benefit patients with various cancers who carry the DDR gene mutation. Cancer chemotherapy plus olaparib or veliparib prolonged the progression-free survival of patients with ovarian cancer who were positive for the BRCA1/2 genes (8, 12), which are DDR genes (13). Moreover, olaparib prolonged the progression-free survival of patients with prostate cancer with any of the 12 DDR gene mutations, including BRCA1/2 (14).
DSB-induced signalling involves non-homologous end joining (NHEJ) DNA repair, which is promoted by p53 binding protein 1 (53BP1) (15). NHEJ is an error-prone DNA repair mechanism that often results in chromosomal aberrations and cell toxicity. Separately, olaparib has been shown to promote the 53BP1-dependent NHEJ pathway, resulting in the accumulation of chromosomal breaks in human bone cell lines (16). In ovarian carcinoma, inhibiting 53BP1 protein expression was associated with attenuation of the anti-tumour effect of PARP inhibitors (17). Knockdown of 53BP1 also decreased the cytotoxicity of PARP inhibitor and DNA crosslinking agents in mammary epithelial cells (18). Because olaparib-induced cytotoxicity is 53BP1-dependent, the synergistic cytotoxicity of anticancer drugs and olaparib may depend on 53BP1 protein expression in OC cell lines.
As such, we performed this study to investigate the role of 53BP1 in the synergistic cytotoxicity of anticancer drugs and olaparib in OC cell lines.
Materials and Methods
Chemicals and reagents. Olaparib was obtained from LKT Laboratories (St. Paul, MN, USA) and 5-FU was purchased from Sigma-Aldrich (St. Louis, MO, USA). CDDP and doxorubicin were purchased from FUJIFILM Wako Pure Chemical Corporation (Osaka, Japan). Docetaxel, SN-38, and temozolomide were purchased from Tokyo Chemical Industry (Tokyo, Japan).
Cell culture. The human OC cell line KYSE70 was obtained from the Health Science Research Resources Bank (Osaka, Japan) (19). Cells were maintained in Dulbecco's modified Eagle's medium (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% heat-inactivated foetal bovine serum (Thermo Fisher Scientific), 100 units/mL of penicillin, and 100 μg/ml of streptomycin (Nacalai Tesque, Kyoto, Japan). Cells were cultured at 37°C in a humidified atmosphere containing 5% CO2.
Transfection of small interfering RNA (siRNA). siRNAs directed at the genes that encode 53BP1and REV7 (also known as mitotic arrest deficient 2 like 2) as well as a negative control (No. 1; siNC) were purchased from Thermo Fisher Scientific. siRNAs were transfected using OptiMEM™ medium (Thermo Fisher Scientific) and Lipofectamine RNAiMAX reagents (Thermo Fisher Scientific) according to manufacturer's instructions. Transfections were performed as described previously (20). In brief, 5×105 cells were plated into 6-well plates and incubated for 24 h, following which they were transfected with siRNA (25 pmol) and incubated for 48 h; the same amount of siRNA was transfected once again for 24 h after which the cells were used for experiments. The siRNA sequences were as follows:
si53BP1: 5’-GAAGGACGGAGUACUAAUATT-3’
siREV7: 5’-CAGACUCGCUGUUGUCUCATT-3’
Western blot analysis. Western blotting was performed as previously described (21). siRNA transfected-KYSE70 cells (2×105) were plated into 6-well plates and incubated for 2 and 6 days. The cells were then harvested and lysed with CelLytic™ M (Sigma-Aldrich) containing 1% protease inhibitor cocktail for use with mammalian cell and tissue extracts (Nacalai Tesque, Kyoto, Japan). Cell lysate protein content was measured using the Bradford method-based protein assay (22) via a commercial reagent purchased from Wako. Equal amounts of protein were loaded on 5-20% (53BP1 and β-actin) or 10-20% (REV7 and β-actin) gradient polyacrylamide gels (SuperSep Ace, FUJIFILM Wako). Following SDS/PAGE separation, proteins were transferred to polyvinylidene difluoride membranes (ClearTrans SP® PVDF membrane, FUJIFILM Wako). Membranes were blocked in phosphate-buffered saline/0.1% Tween 20 (PBS-T) containing 1% skim milk (FUJIFILM Wako) and incubated with primary antibodies at 4°C overnight. The following primary antibodies were used: anti-53BP1 rabbit monoclonal IgG [EPR2172(2), Abcam, Cambridge, UK], anti-REV7 rabbit monoclonal IgG (EPR13657, Abcam), and anti-β-actin mouse monoclonal IgG1 (2F3, FUJIFILM Wako). The anti-53BP1 (1:4,000) or anti-REV7 (1:4,000) antibody was diluted in Can Get Signal Immunoreaction Enhancer Solution I (Toyobo, Osaka, Japan), while the anti-β-actin (1:1,000) antibody was diluted in PBS-T. Secondary horseradish peroxidase-conjugated antibodies were incubated with the membranes for 1 h at room temperature. The following secondary antibodies were used: anti-rabbit IgG antibodies (GE Healthcare, Little Chalfont, UK) for REV7 or 53BP1, and anti-mouse IgG antibodies (GE Healthcare) for β-actin. The anti-rabbit (1:20,000) and anti-mouse (1:25,000) antibodies were diluted in Can Get Signal Immunoreaction Enhancer Solution II and PBS-T, respectively. Proteins were detected using the ImmunoStar® LD reagent (FUJIFILM Wako) and visualized with Fusion Solo S (Vilber-Lourmat, Collégien, France).
Growth inhibition assay. Cell survival was measured using the CellQuanti-Blue™ Cell Viability Assay Kit (BioAssay Systems, Hayward, CA, USA) as described previously (18). In brief, siRNA transfected-KYSE70 cells (1×103) were seeded into a 96-well plate for 24 h. Cells were treated for 1 week to various concentrations of anticancer drugs with or without olaparib (5 μM). After the medium was removed, cells were treated with CellQuanti-Blue™ reagent solution (medium:reagent=10:1). After 5 h, the fluorescence intensity was measured using a PowerScan® HT multi-mode microplate reader (DS Pharma Biomedical, Osaka, Japan) at excitation and emission wavelengths of 535 and 590 nm, respectively. Half-maximal inhibitory concentrations (IC50 values) were calculated according to the sigmoid inhibitory effect model (eq. 1) using the nonlinear least-squares fitting method (Solver, Microsoft Excel; Redmond, WA, USA).
 (1)
(1)E and Emax represent the surviving fraction (% of control) and its maximum, respectively. C and γ are the drug concentration in the medium and the sigmoidicity factor, respectively (23).
Immunofluorescence analysis. KYSE70 cells (2×104) were grown in black 96-well plates for 24 h. Cells were treated with anticancer drugs with or without olaparib (5 μM) for 24 h. The cells were then treated for 10 min with 0.2% Triton X-100 in PBS on ice for REV7 and 53BP1 co-detection but not for phosphorylated H2A histone family member X (γH2AX: a biomarker of DSBs) (24) and 53BP1 co-detection. Next, cells were fixed with 4% paraformaldehyde for 15 min, permeabilised with 0.2% Triton X-100 in PBS for 30 min, and blocked for 1 h in PBS containing 0.1% bovine serum albumin. The cells were then incubated overnight with primary antibodies at 4°C. The primary antibodies except for those conjugated to a fluorescent label were probed with secondary antibodies for 1 h at room temperature, after which the fluorescent-labelled primary antibody against the co-detected protein was incubated at 4°C overnight. The primary and secondary antibodies used for γH2AX and 53BP1 co-detection and their dilutions were as follows: anti-phospho-histone H2AX Ser139 (γH2AX) mouse monoclonal IgG1 (JBW301, 1: 20,000; Merck Millipore, Billerica, MA, USA); anti-53BP1 rabbit monoclonal IgG antibody [EPR2172(2), 1: 20,000, Abcam]; anti-mouse IgG (H/L), F(ab’) 2 Fragment Alexa Fluor® 488 Conjugate (1:1,000, Cell Signalling Technology, Danvers, MA, USA) for γH2AX-detection; and anti-rabbit IgG (H+L), F(ab’)2 Fragment Alexa Fluor® 647 Conjugate (1:1,000, Cell Signalling Technology) for 53BP1-detection. For REV7 and 53BP1 co-detection, the primary or secondary antibodies and their dilutions were as follows: anti-53BP1 rabbit monoclonal IgG antibody Alexa Fluor® 488 conjugate [EPR2172(2), 1: 20,000; Abcam]; anti-REV7 rabbit monoclonal IgG (EPR13657, 1: 20,000; Abcam); and anti-rabbit IgG (H+L), F(ab’)2 Fragment Alexa Fluor® 647 Conjugate (1:1,000, Cell Signalling Technology) for REV7-detection. All primary and secondary antibodies were diluted in Can Get Signal Immunostain Solution A (Toyobo) and PBS-T, respectively. Cell nuclei were stained with 0.2 μg/ml 4’,6-diamidino-2-phenylindole (DAPI) in PBS. Cells were visualized using an Operetta High Content imaging microscope (PerkinElmer, Waltham, MA, USA). The numbers and co-localisations of nuclear foci were analysed by recognizing Alexa Fluor® 488 (γH2AX or 53BP1) or Alexa Fluor® 647 (53BP1 or REV7) spots using the Harmony software (PerkinElmer). At least 100 cells per sample were analysed.

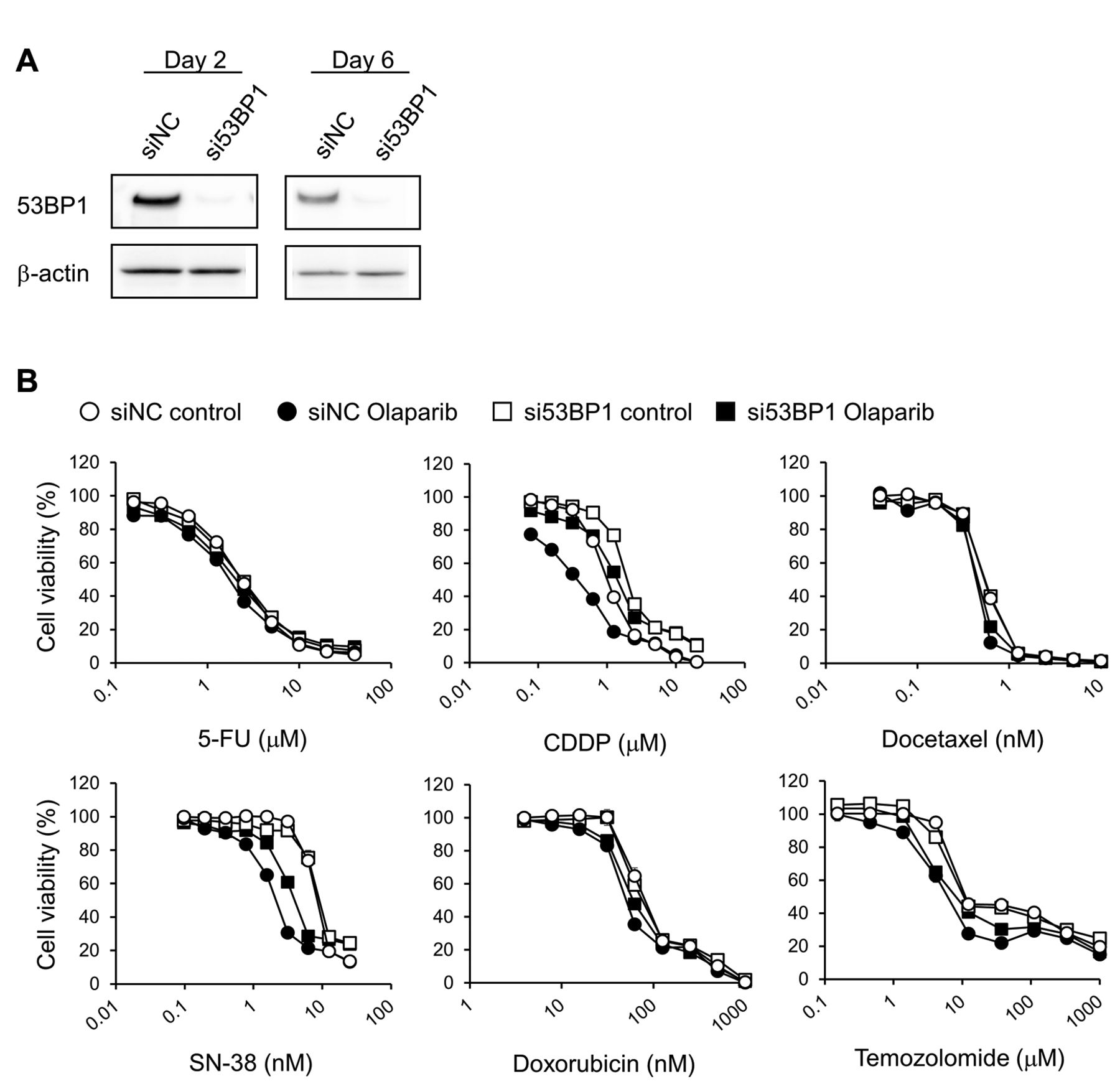
Effects of si53BP1-transfection on the cytotoxicity of anticancer drugs administered with or without olaparib to KYSE70 cells. (A) Two and 6 days after transfection, total protein was extracted from whole-cell lysates, and western blotting was performed for 53BP1 and β-actin (reference protein) detection. (B) siRNA-transfected cells were seeded onto 96-well plates. After culturing for 24 h, cells were exposed continuously to the indicated drug concentrations for 1 week with or without olaparib (5 μM). Cell viability was determined using the CellQuanti-Blue™ cell viability assay kit. Each point represents the mean±standard error (n=4). siRNA, Small interfering RNA; si53BP1, siRNA against 53BP1; siNC, negative control siRNA; 5-FU, 5-fluorouracil; CDDP, cisplatin; SN-38, 7-ethyl-10-hydroxy-camptothecin.
Apoptosis assay. siRNAs treated-KYSE70 cells (2.0×106) were seeded onto 6-well plates and incubated for 24 h. The cells were treated with anticancer drugs with or without olaparib (5 μM) for 48 h. The floating and adherent cells were harvested and suspended in annexin V binding buffer [10 mM HEPES/NaOH (pH 7.4), 140 mM NaCl and 2.5 mM CaCl2]. Next, the cells were stained with annexin V-fluorescein isothiocyanate (BioLegend, SanDiego, CA, USA) and propidium iodide (50 μg/ml, Wako) for 15 min. Cellular fluorescence was detected using the LSRFortessa™X-20 flow cytometer (Becton Dickinson, San Jose, CA, USA), and analysed using the FACSDiva™ software (Becton Dickinson). At least 10,000 cells per sample were analysed.
IC50 values of anticancer drugs without or with olaparib in siNC- or si53BP1-transfected KYSE70 cells.
IC50 values of anticancer drugs without or with olaparib in siNC- or siREV7-transfected KYSE70 cells.
Statistical analyses. All statistical analyses were performed using EZR (25). Data are presented as means±standard error. Experiments with 3 or more groups were assessed using a repeated one-way analysis of variance followed by Tukey's honest significant difference test. All analyses were conducted with 2-tailed p-values and considered statistically significant when p<0.05.
Results
53BP1 knockdown attenuates the cytotoxicity of anticancer drugs plus olaparib in KYSE70 cells. The expression of 53BP1 was almost completely suppressed at 2 and 6 days after si53BP1 transfection (Figure 1A). Olaparib shifted the growth inhibitory curves of all anticancer drugs except 5-FU to lower concentrations, and significantly decreased their IC50 values, in siNC-transfected cells (Figure 1B and Table I). The transfection of si53BP1 into KYSE70 cells shifted the growth inhibitory curves of CDDP and SN-38 with or without olaparib to higher concentrations, and significantly increased the IC50 values of CDDP and SN-38 with or without olaparib relative to siNC-transfected cells. The ratios of each cytotoxic drug's IC50 value in si53BP1-transfected cells to that in siNC-transfected cells (si53BP1/siNC IC50 ratio) were higher with olaparib (CDDP+olaparib, 3.25; SN-38+olaparib, 2.12) than without (CDDP alone, 2.07; SN-38 alone, 1.19) (Figure 1B and Table I). The transfection of si53BP1 did not shift the growth inhibitory curve and IC50 value of doxorubicin in the absence of olaparib, although temozolomide curves without olaparib shifted to lower concentrations and lower IC50 values following si53BP1 transfection. However, the growth inhibitory curves of doxorubicin and temozolomide with olaparib shifted to higher concentrations and increased IC50 values following si53BP1 transfection. The si53BP1/siNC IC50 ratios of doxorubicin+olaparib (1.19) and temozolomide+olaparib (1.65) were higher than those of either drug alone (0.98 and 0.42, respectively). However, si53BP1 transfection hardly affected the growth inhibitory curves and IC50 values of 5-FU and docetaxel irrespective of olaparib treatment (Figure 1B and Table I).

Effects of olaparib on the nuclear accumulation of γH2AX and 53BP1 induced by anticancer drugs in KYSE70 cells. Cells were seeded onto black 96-well plates. After culturing for 24 h, cells were exposed continuously to the indicated anticancer drug with or without olaparib (5 μM) for 24 h. The concentrations of anticancer drugs are as follows: (A) 5-FU, 1 μM; (B) CDDP, 1 μM; (C) docetaxel, 1 nM; (D) SN-38, 10 nM; (E) doxorubicin, 100 nM; and (F) temozolomide, 100 μM. Cells were immunostained for γH2AX or 53BP1, and their accumulation in the nucleus was examined by immunofluorescence microscopy after counterstaining with DAPI. Each set of panels shows the numbers of γH2AX foci in the nucleus (left) and the numbers of 53BP1 foci co-localised with γH2AX (right). Each bar represents the mean±standard error (n=3). Significant differences were determined by an analysis of variance followed by Tukey's test (**p<0.01 vs. control †p<0.05, ††p<0.01, N.S., not significant). 5-FU, 5-Fluorouracil; CDDP, cisplatin; SN-38, 7-ethyl-10-hydroxy-camptothecin.

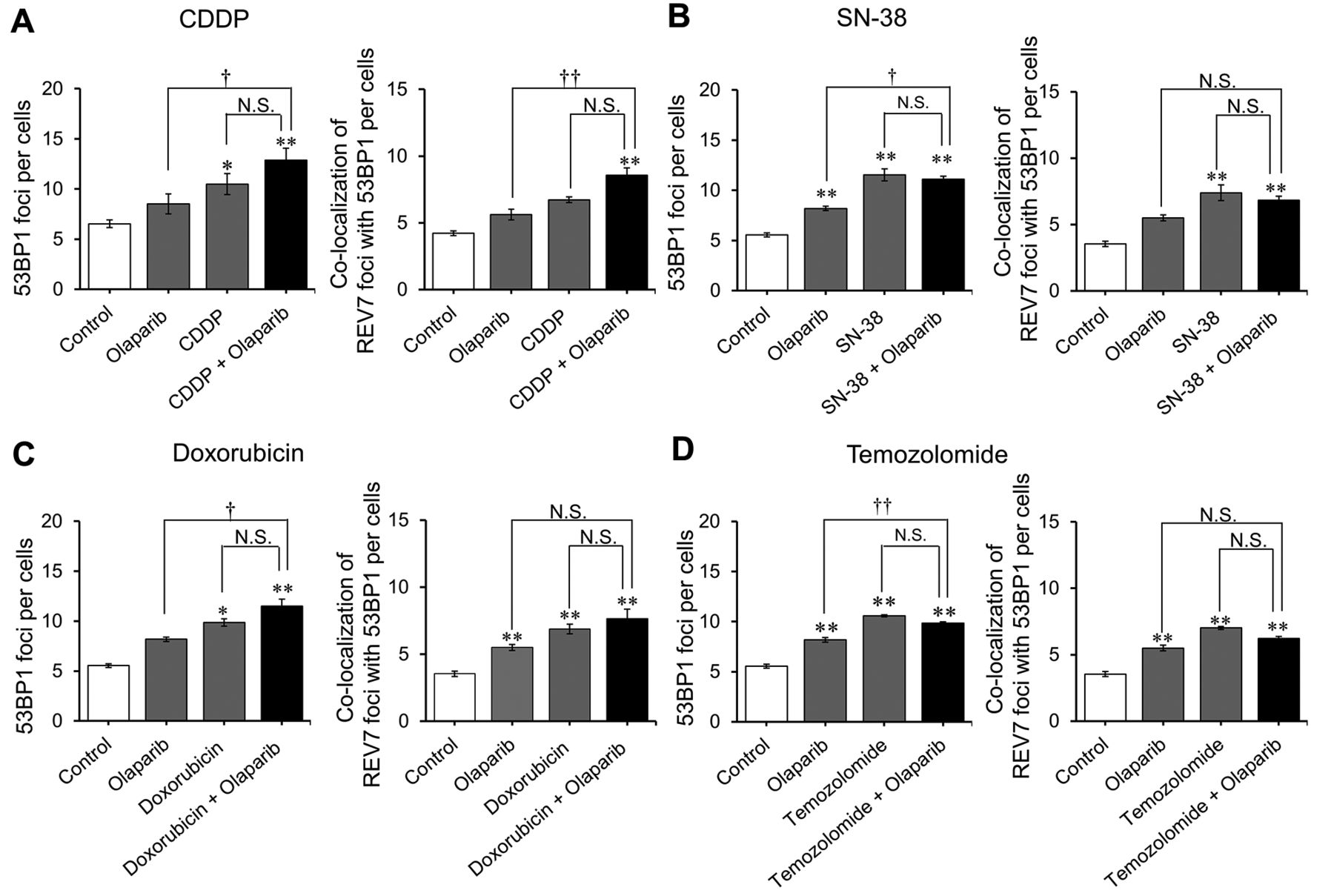
Effects of olaparib on the nuclear accumulation of 53BP1 and REV7 caused by anticancer drugs in KYSE70 cells. Cells were seeded onto black 96-well plates. After culturing for 24 h, cells were exposed continuously to the indicated anticancer drug with or without olaparib (5 μM) for 24 h. The concentrations of anticancer drugs are as follows: (A) CDDP, 1 μM; (B) SN-38, 10 nM; (C) doxorubicin, 100 nM; and (D) temozolomide, 100 μM. Cells were immunostained for 53BP1 or REV7, and their accumulations in the nucleus were examined by immunofluorescence microscopy after counterstaining with DAPI. Each set of panels shows the number of 53BP1 foci in nucleus (left) and the number of REV7 foci co-localised with 53BP1 (right). Each bar represents the mean±standard error (n=3). Significant differences were determined by an analysis of variance followed by Tukey's test (*p<0.05; **p<0.01 vs. control; †p<0.05; ††p<0.01; N.S., not significant). 5-FU, 5-Fluorouracil; CDDP, cisplatin; SN-38, 7-ethyl-10-hydroxy-camptothecin.
Effects of olaparib on drug-induced nuclear accumulation of γH2AX and 53BP1 in KYSE70 cells. Olaparib increased the number of γH2AX foci and the amount of 53BP1-γH2AX co-localisation. CDDP, SN-38, doxorubicin, and temozolomide also increased these numbers in KYSE70 cells, while 5-FU and docetaxel did not. Olaparib additively increased the number of γH2AX foci and induced 53BP1-γH2AX co-localisation in CDDP-, doxorubicin-, SN-38- and temozolomide-treated cells (Figure 2).
Effects of olaparib on drug-induced nuclear accumulation of 53BP1 and REV7 in KYSE70 cells. CDDP, SN-38, doxorubicin, and temozolomide increased the accumulation of 53BP1 foci as well as of REV7 foci co-localised with 53BP1 in KYSE70 cells. However, olaparib inclusion with these treatments did not produce an additive increase in the same (Figure 3A-D).
Knockdown of REV7 increased the cytotoxicity of anticancer drug with and without olaparib in KYSE70 cells. REV7 protein expression was suppressed almost completely at 2 and 6 days post-siREV7 transfection (Figure 4A). Transfection of siREV7 shifted the growth inhibitory curves of CDDP, doxorubicin, SN-38, and temozolomide to lower concentrations with or without olaparib treatment; the IC50 values of these drugs were significantly decreased following siREV7 transfection with or without olaparib (Figure 4B and Table II).

Effects of siREV7-transfection on the cytotoxicity of anticancer drug with or without olaparib in KYSE70 cells. (A) After transfection for 2 and 6 days, total protein was extracted from whole-cell lysates, and western blotting was performed for REV7 and β-actin (reference protein) detection. (B) siRNA-transfected cells were seeded onto 96-well plates. After culturing for 24 h, cells were exposed continuously to the indicated drug concentrations with or without olaparib (5 μM) for 1 week. Cell viability was determined using the CellQuanti-Blue™ cell viability assay kit. Each point represents the mean±standard error (n=4). siRNA, Small interfering RNA; siREV7, siRNA against REV7; siNC, negative control siRNA; 5-FU, 5-fluorouracil; CDDP, cisplatin; SN-38, 7-ethyl-10-hydroxy-camptothecin.
Effects of 53BP1 silencing on anticancer drug-induced KYSE70 cell apoptosis with or without olaparib. The anticancer drugs significantly increased the number of annexin V-positive cells in siNC-transfected cells; however, olaparib did not produce an additional increase in the same. In si53BP1-transfected cells, no increase in the number of annexin V-positive cells was observed following anticancer drug and olaparib treatment (Figure 5A-D).
Discussion
Olaparib enhanced the sensitivity of anticancer drugs (other than 5-FU) in KYSE70 cells. Depletion of 53BP1 attenuated the sensitivity of CDDP and SN-38 without olaparib in KYSE70 cells. In the presence of olaparib, the sensitivities of CDDP, SN-38, doxorubicin, and temozolomide (which are DNA damaging agents) were attenuated following 53BP1 silencing; this attenuation was greater with olaparib co-treatment. However, depletion of REV7, a downstream effector of 53BP1-dependent NHEJ repair, did not attenuate the sensitivity of KYSE70 cells to anticancer drugs irrespective of olaparib treatment. These findings indicate that 53BP1 promotes the cytotoxicity of olaparib as well as its sensitizing of OC cells to DNA synthesis inhibitors in a manner that is independent of the 53BP1/REV7 pathway.
Olaparib induced DNA damage in CDDP-, SN-38-, doxorubicin-, and temozolomide-treated KYSE70 cells, and also increased the number of 53BP1 foci co-localised with γH2AX in anticancer drugs-treated cells. In a previous study, SN-38 plus olaparib produced an additive increase in the number of nuclear γH2AX and 53BP1 foci in colon cancer cell lines (26). TP53BP1 with a Phe1553Arg-mutation, a hyperactive form of 53BP1 involved in DDR, enhanced the cytotoxicity of olaparib in mouse embryonic fibroblasts (27). Hence, the sensitivity of CDDP, SN-38, doxorubicin, and temozolomide may be increased by olaparib via the accumulation of 53BP1 protein on damaged DNA sites in OC cell lines.

Effects of si53BP1 transfection on KYSE70 cell apoptosis caused by anticancer drugs with and without olaparib. siRNA-transfected cells were seeded onto 6-well plates. After culturing for 24 h, cells were exposed continuously to the indicated anticancer drug with or without olaparib (5 μM) for 48 h. The concentrations of anticancer drugs were as follows: (A) CDDP, 1 μM; (B) SN-38, 10nM; (C) doxorubicin, 100 nM; and (D) temozolomide, 100 μM. Next, cells were harvested and stained with annexin V-fluorescein isothiocyanate and propidium iodide, and analysed using the Becton Dickinson LSRFortessa™ instrument. Each bar represents the mean±standard error (n=3). Significant differences were determined via analysis of variance followed by Tukey's test (*p<0.05; **p<0.01 vs. control of siNC; †p<0.05; and ††p<0.01). siRNA, Small interfering RNA; si53BP1, siRNA against 53BP1; siNC, negative control siRNA; CDDP, cisplatin; SN-38, 7-ethyl-10-hydroxy-camptotheci.
Olaparib did not increase the number of REV7 foci co-localised with 53BP1 in anticancer drug-treated cells. Moreover, depletion of REV7 did not attenuate the cytotoxicity of anticancer drugs regardless of olaparib presence. After DSBs are induced, 53BP1 recruits the shieldin complex that includes REV7 to the damaged DNA (24); in the presence of olaparib, this complex promotes the error-prone classical NHEJ (c-NHEJ) repair pathway and induced cell death by introducing chromosome aberrations (28, 29). However, the mechanism by which cytotoxic anticancer drugs synergize with olaparib may be separate from that which introduces chromosomal aberrations through the 53BP1/REV7 pathway, although both mechanisms may still influence the 53BP1-dependent DDR pathway.
Olaparib induced apoptosis and cell cycle arrest through the accumulation of nuclear γH2AX and phosphorylated ‘ataxia telangiectasia mutated’ (ATM) in breast cancer cell lines; however, 53BP1 silencing reversed olaparib-induced apoptosis and cell cycle arrest (30). DNA damage induced by irradiation increased ATM via the expression of pro-apoptotic genes through p53 or p73 modulation as well as cell cycle arrest via p27 (30-34). However, in our study, olaparib did not increase anticancer drug-induced apoptosis in OC cell lines, and 53BP1 depletion did not increase apoptosis regardless of olaparib treatment. These results suggest that the synergistic cytotoxicity of anticancer drugs and olaparib is not a result of 53BP1/ATM/p53/p73-mediated apoptosis.
In the presence of olaparib, the sensitivity of CDDP, SN-38, doxorubicin, and temozolomide were attenuated following 53BP1 silencing; however, the drug-sensitizing effects of olaparib were not attenuated by REV7 silencing in KYSE70 cells. The 53BP1 protein is known to promote DNA resection-independent (i.e., shieldin complex-dependent) c-NHEJ and DNA resection-dependent microhomology-mediated end joining (MMEJ, which is a type of Artemis-dependent NHEJ repair) (35, 36). MMEJ is known as a highly mutagenic DSB repair pathway because it often causes several base insertions or deletions (37). Ito et al. (38) reported that olaparib potentiated the cytotoxicity of CDDP via accumulation of chromosome aberration in human mammary epithelium cell line. These results suggest that olaparib activates MMEJ pathway caused by DNA damaging agents in KYSE70 cells. Hence, olaparib may increase 53BP1-dependent mutagenic MMEJ repair of DNA damage induced by anticancer drugs plus olaparib, and thereby enhance the sensitivity of DNA-damaging agents in OC cell lines.
In conclusion, our data suggest that the cytotoxicity of CDDP, SN-38, doxorubicin, and temozolomide is increased by olaparib via an 53BP1-related mechanism in OC cell lines. This mechanism may not be dependent on c-NHEJ repair via the 53BP1/REV7 pathway or on 53BP1-dependent apoptosis, but may rather depend on 53BP1-mediated DDR pathway activity.
Acknowledgements
This study was supported in part by the JSPS KAKENHI (grant number: 16K18964).
Footnotes
Authors' Contributions
Study concept and design: T. Minegaki, K. Miyamoto. Acquisition of data: K. Miyamoto, T. Minegaki, S. Hirano, I. Hayashi. Analysis and interpretation of data: all authors. Drafting of the manuscript: K. Miyamoto, T. Minegaki. Study supervision: M. Tsujimoto, K. Nishiguchi.
Conflicts of Interest
The Authors declare no conflicts of interest associated with this manuscript.
- Received December 19, 2019.
- Revision received January 10, 2020.
- Accepted January 14, 2020.
- Copyright© 2020, International Institute of Anticancer Research (Dr. George J. Delinasios), All rights reserved
References
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Therapeutic Options Targeting the Ataxia-Telangiectasia Mutated (ATM)-mediated DNA Damage Response, Macropinocytosis, and Adaptive Immunity in Ovarian Cancer
- Circular RNAs With Efficacy in Preclinical In Vitro and In Vivo Models of Esophageal Squamous Cell Carcinoma
- Detection of Endogenous DNA Double-strand Breaks in Oral Squamous Epithelial Lesions by P53-binding Protein 1
- Induction of Cross-resistance to ABCB1 Substrates in Venetoclax-resistant Human Leukemia HL60 Cells