


Abstract
Background/Aim: Aberrant expression of the SEI1 oncogene has been prevalently found in a variety of human cancers, including oral squamous cell carcinoma (OSCC). Recent studies have shown that cisplatin up-regulates the expression of SEI1 in breast and bladder cancer cells, thus inhibiting apoptosis and cell death in these cells. In the present study, we investigated the impact of cisplatin on the expression of SEI1 in OSCC cells. Materials and Methods: Four OSCC cell lines, CAL27, SCC4, SCC15, and SCC22A were treated with cisplatin and 5-fluorouracil, and changes in SEI1 expression in these cells were evaluated using quantitative Reverse Transcription-Polymerase Chain Reaction (qRT-PCR) analyses. Results: Cisplatin significantly induced SEI1 expression in the tested OSCC cells. Contrarily, cisplatin treatment did not affect the expression of gankyrin and BMI1, two oncogenes frequently overexpressed in a coordinate manner with SEI1 in OSCC. Additionally, 5-fluorouracil did not bring about any detectable changes in SEI1 expression in these cells. Conclusion: Cisplatin-induced up-regulation of SEI1 expression in OSCC is specific, and such induction could underlie the development of resistance to cisplatin in OSCC.
Oral cancer is an important global health concern accounting for more than 550,000 cases and 380,000 deaths annually worldwide and is the 6th most common cancer type (1). The majority (>90%) of oral cancer cases are oral squamous cell carcinomas (OSCC) (2-4). Despite advances in diagnostic techniques and improvements in treatment modalities in the past decades, the 5-year survival rate of patients with OSCC (<60%) has not improved (5). To date, chemotherapy, radiation, and surgery remain the standard care for OSCC (6). First approved by the U.S. Food and Drug Administration in 1978 for its use in testicular and ovarian cancers, cisplatin (as well as other platinum-based analogs) has been one of the most commonly used chemotherapeutic agents in patients with a wide variety of cancers including OSCC (7). While cisplatin delivers initial success with partial responses and disease stabilization in many patients (8, 9), its clinical use is ultimately compromised due to its side effects and the development of resistance to cisplatin in patients. Knowledge of the molecular mechanisms underlying the development of resistance to cisplatin in OSCC and other cancers remains limited.
It is well known that the SEI1 (TRIP-Br1) gene product, p34SEI1, exerts oncogenic effects via regulation of the cell cycle, apoptosis, senescence, and autophagy (10-19). P34SEI1 specifically binds to the cyclin D-dependent kinase 4 (CDK4)/cyclin D1/p16INK4A complex and diminishes the inhibition of p16INK4A on the kinase activity (10). P34SEI1 also possess an intrinsic transactivation activity and regulates the transcriptional activity of E2F-1 via interaction with DP-1, an E2F-1 partner protein, thus modulating the expression of genes required for cell cycle progression, such as cyclin E (11, 12). Furthermore, p34SEI1 inhibits apoptosis through 1) binding to the X-linked inhibitor of apoptosis protein, XIAP, and protecting the latter from degradation (13-15), 2) modulating p53-dependent transcriptional activation (16), and 3) down-regulating the tumor suppressor PTEN though NEDD-1-mediated PTEN ubiquitination/degradation (17-19). Previous studies in our laboratory and from other groups have shown that SEI1 is prevalently overexpressed in OSCC, esophageal, breast, ovarian, brain, liver, and lung cancers (12, 15, 20, 21). A recent meta-analysis showed that SEI1 overexpression significantly reduced the median overall-survival of patients with liver and ovarian cancers (21). Interestingly, it has been reported that up-regulation of SEI1 in breast cancer cells inhibited hypoxia-induced apoptosis and autophagy, thus providing cancer cells resistance to the hypoxia-induced cell death (22). In addition, it has been demonstrated that cisplatin treatment induced up-regulation of SEI1 in bladder cancer cells regardless of the TP53 gene status (23, 24). With regard to all these findings, we postulated that up-regulation of SEI1 might underlie the development of resistance to cisplatin, an apoptosis-induced agent, in OSCC. In the present study, we evaluated the change in SEI1 mRNA expression in four OSCC cell lines upon cisplatin treatment, and our results showed that cisplatin significantly induced SEI1 expression in all tested OSCC cells. In contrast, cisplatin treatment did not impact the expression of gankyrin and BMI1, two oncogenes frequently overexpressed in a coordinate manner with SEI1 in OSCC specimens (20). Interestingly, 5-fluorouracil (5FU), another chemotherapeutic agent widely used in the chemotherapy of OSCC (25), did not bring about significant changes in SEI1 expression in the tested OSCC cell lines. These results indicate that cisplatin-induced up-regulation of SEI1 is specific in OSCC, and such induction could underlie the development of resistance to cisplatin in OSCC.
Materials and Methods
Cell lines and reagents. CAL27, SCC4 and SCC15 were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA); SCC22A was a kind gift from Dr. Christopher Weghorst at The Ohio State University School of Public Health. All cell lines were maintained in advanced Dulbecco modified Eagle medium (DMEM)/Ham F12 medium ((Life Technologies Corporate, Carlsbad, CA, USA) supplemented with 5% fetal bovine serum (FBS; Life Technologies), 1% glutamine (Life Technologies), 1% penicillin/streptomycin (Life Technologies) at 37°C, in a humidified atmosphere with 5% CO2. These cell lines were regularly authenticated using short tandem repeat polymorphism (STRP) analysis as recommended by ATCC, and were mycoplasma free. Cells were grown up to passage 20. Cisplatin and 5FU were purchased from Cayman Chemicals, Inc. (Ann Arbor, MI, USA).
Cell viability assay. Cells were seeded at 2000 cells/well in 100 μl of advanced DMEM/F12-5% FBS media and incubated at 37°C and 5% CO2 overnight. Subsequently, cells were incubated with media containing various concentrations of cisplatin or 5FU for another 24 h. Cell viability was assayed using WST-1 Cell proliferation Assay kit (Roche, Indianapolis, IN, USA) following the manufacturer's directions. Assays were performed in triplicate at least twice. Absolute IC50 values (the concentration of cisplatin required to inhibit 50% of the cell viability) were determined using Kaleidagraph software (Synergy Software, Reading, PA, USA) as previously described (26).
Gene expression assay. Cells were seeded at 1×106 cells/T25 flask (Life technologies) and incubated in DMEM/F12-10% FBS media overnight. Cells were then incubated with media containing cisplatin or 5-FU at the indicated concentrations (cisplatin: 0 μM, 6.6 μM, 10 μM, 20 μM, and 30 μM; 5FU: 0 μM, 25 μM, 50 μM, 100 μM, and 200 μM) at 37°C and 5% CO2 for another 24 h. Cells were harvested by centrifugation and total RNA was purified using a RNeasy Purification kit (Qiagen, Valencia, CA, USA). cDNAs were synthesized using a High Capacity cDNA Reverse Transcription kit (Life Technologies). The expression levels of target genes were quantitatively assessed using Taqman® gene expression assays (Life Technologies) using the following inventoried primer/probes: Hs00175935_m1 for CDK4, Hs00277039_m1 for cyclin D1, Hs0023356_m1 for cyclin E, Hs00829508-s1 for gankyrin, Hs00203547_m1 for SEI1, Hs00409825_g1 for BMI1, Hs00355782_m1 for p21 (CDKN1A), Hs00154374_m1 for CDC6, and Hs99999909_m1 for human hypoxanthine phosphoribosyl-transferase (HPRT1). Of note, HPRT1 was used as an endogenous reference for normalized gene expression. Cisplatin treatment experiments were performed in triplicate. For each cDNA sample, target genes were amplified separately, and expression quantitation assays were performed in triplicate. The relative gene expression level (REL) of a target gene was determined using a comparative Cq method in which REL was defined as 2-ΔCq. Change in the expression of a target gene in a cell line after cisplatin treatment was defined as 2−ΔΔCq, i.e. the ratio of REL with the indicated concentration of cisplatin and REL without cisplatin. A change of ≥2-fold was regarded as significant in gene expression (27).
Results
Cisplatin inhibited the growth of OSCC cells. As an alkylating agent, cisplatin covalently binds to DNA bases and forms DNA crosslink adducts, which interfere with the DNA repair machinery and trigger apoptosis (7-9). In this study, we first evaluated the inhibitory ability of cisplatin in four selected OSCC cell lines, namely, CAL27, SCC4, SCC15, and SCC22A. As shown in Figure 1A, cisplatin exhibited considerable inhibitory activity in these cell lines. Under experimental conditions (with 24-h treatment), the IC50 values of cisplatin, i.e. the concentrations required to inhibit 50% of cell viability, were 11.8±0.7 μM, 28.5±1.3 μM, 20.2±1.4 μM, and 24.3±2.0 μM in CAL27, SCC4, SCC15, and SCC22A, respectively. While SCC4, SCC15, and SCC22A cell lines had comparable IC50 values, CAL27 appeared to be more sensitive to cisplatin than the other three cell lines. Notably, all these four OSCC cell lines harbored TP53 mutations, and one of them, SCC22A, even had nonfunctional p53 protein (28, 29). The aforementioned results indicate that to a certain extent, cisplatin inhibition in OSCC cell lines is independent of TP53 status. This observation is consistent with previous studies showing that cisplatin inhibits cell cycle progression and/or induces apoptosis in cells in both p53-dependent and p53-independent manners (23, 24, 30-32).
Cisplatin induced SEI1 up-regulation in OSCC cells. We then evaluated the expression of SEI1 in these OSCC cells upon treatment with different concentrations of cisplatin for 24 h. As shown in Figure 2A, cisplatin, at relatively high concentrations (20 μM and 30 μM), significantly increased the expression of SEI1 in all tested cells. At the concentration of 20 μM, cisplatin induced an increase in SEI1 expression by an average of 6.83-fold (standard deviation: 1.94); at the concentration of 30 μM, cisplatin induced an increase in SEI1 expression by an average of 12.88-fold (standard deviation: 2.11). Even at low concentrations (6.6 μM and 10 μM), cisplatin was able to up-regulate SEI1 expression in all tested OSCC cell lines except SCC15. Overall, higher concentrations of cisplatin tended to induce higher expression of SEI1 in the tested OSCC cells.

Effect of cisplatin (A) and 5FU (B) on the growth of different oral squamous cell carcinoma (OSCC) cell lines. OSCC cells were incubated with various concentrations of cisplatin or 5FU for 24 h. Cell viabilities were then evaluated using WST-1 cell proliferation Assay kit following the manufacturer's instruction, and IC50 values, the concentrations of cisplatin required to inhibit 50% of the cell viability, were calculated using a 4-parameter non-linear regression approach. Experiments were conducted at least in triplicate.
Previous studies have shown that the intrinsic transactivation activity of p34SEI1 is able to modulate the E2F-related and p53-related transcription, thus up-regulating the expression of cyclin E and p21 in cells, respectively (23, 24). Hence, we continued to evaluate the expression of cylcin E and p21 in these OSCC cells upon cisplatin treatment. Our results showed that cisplatin, at the high concentration (30 μM), induced considerable increase (>2-fold) in the expression of cylcin E in all tested SOCC cells (Figure 2B). Additionally, high concentrations of cisplatin (20 μM and 30 μM) up-regulated the expression of p21 in three tested cell lines, CAL27, SCC4, and SCC22A (Figure 2C). As for SCC15, cisplatin at the low concentration of 6.6 μM led to an >2-fold increase in p21 expression, whereas the changes in p21 expression in SCC15 caused by high concentrations of cisplatin were around 2-fold.
Cisplatin did not impact the expression of gankyrin and BMI1 in OSCC cells. It has been also reported that some oncogenes, such as SEI1, cyclin E, gankyirin, BMI1, cyclin D1, CDK4, are up-regulated in a coordinate manner in oral cancer progression (20). While our results showed that SEI1, cyclin E, and p21 were up-regulated in OSCC cells upon cisplatin treatment, we subsequently investigated the potential impacts of cisplatin on the expression of gankyrin, BMI1, cyclin D1, and CDK4 in these cells. Our results demonstrated that cisplatin treatment led to no detectable change in the expression of gankyrin (Figure 3A), BMI1 (Figure 3B), and CDK4 (data now shown) in these cells. As for cyclin D1, its expression was not up-regulated in any of these OSCC cell lines (Figure 3C). In contrast, down-regulation of cyclin D1 expression was observed in CAL27 and SCC4 in the presence of cisplatin (10 μM, 20 μM, and 30 μM). Taken together, these results indicate that cisplatin-induced up-regulation of SEI1 expression in OSCC cells is gene-specific.
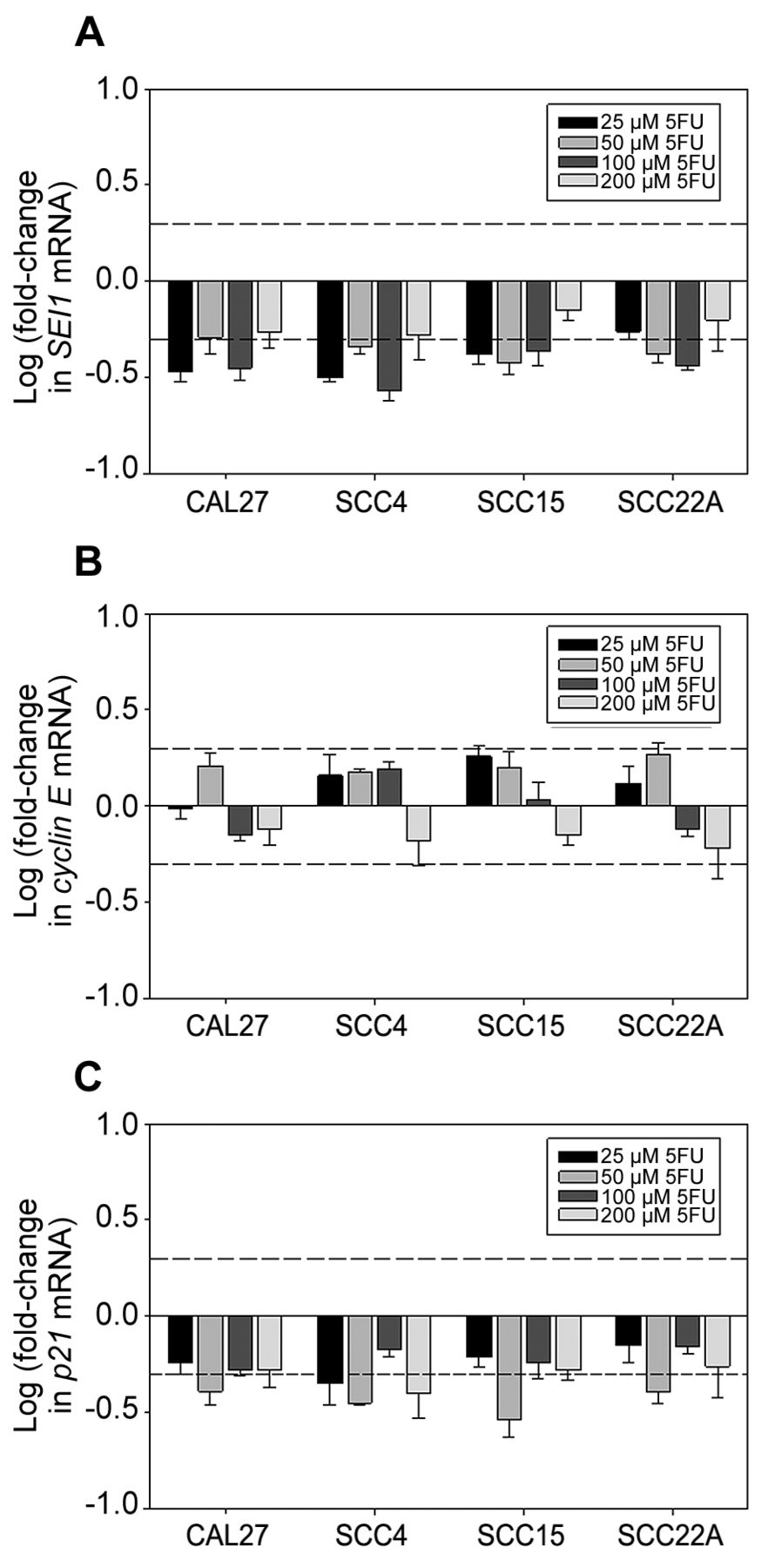
5FU did not impact the expression of SEI1 in OSCC cells. To further investigate the specificity of cisplatin-induced SEI1 up-regulation in OSCC cells, we evaluated the impact of 5FU on the expression of SEI1 in these cells. 5FU is another chemotherapeutic agent widely used in cancer therapy (25), which acts as a suicide inhibitor of thymidylate synthase that inhibits DNA synthesis and replication. As shown in Figure 1B, 5FU exhibited comparable inhibitory activities in all four tested OSCC cell lines with IC50 values ranging from 100 to 200 μM. Interestingly, 5FU (ranging from 25 μM to 200 μM) did not bring about any detectable increase in the expression of SEI1 in OSCC cells (Figure 4A). In contrast, 5FU appeared to down-regulate the expression of SEI1 in these cells. Accordingly, no up-regulation in cyclin E expression (Figure 4B) nor p21 expression (Figure 4C) was observed in these cells. Contrarily, p21 expression tended to be down-regulated in these cells upon 5FU treatment. Overall, these results imply that cisplatin-induced up-regulation of SEI1 expression tends to be drug-specific.
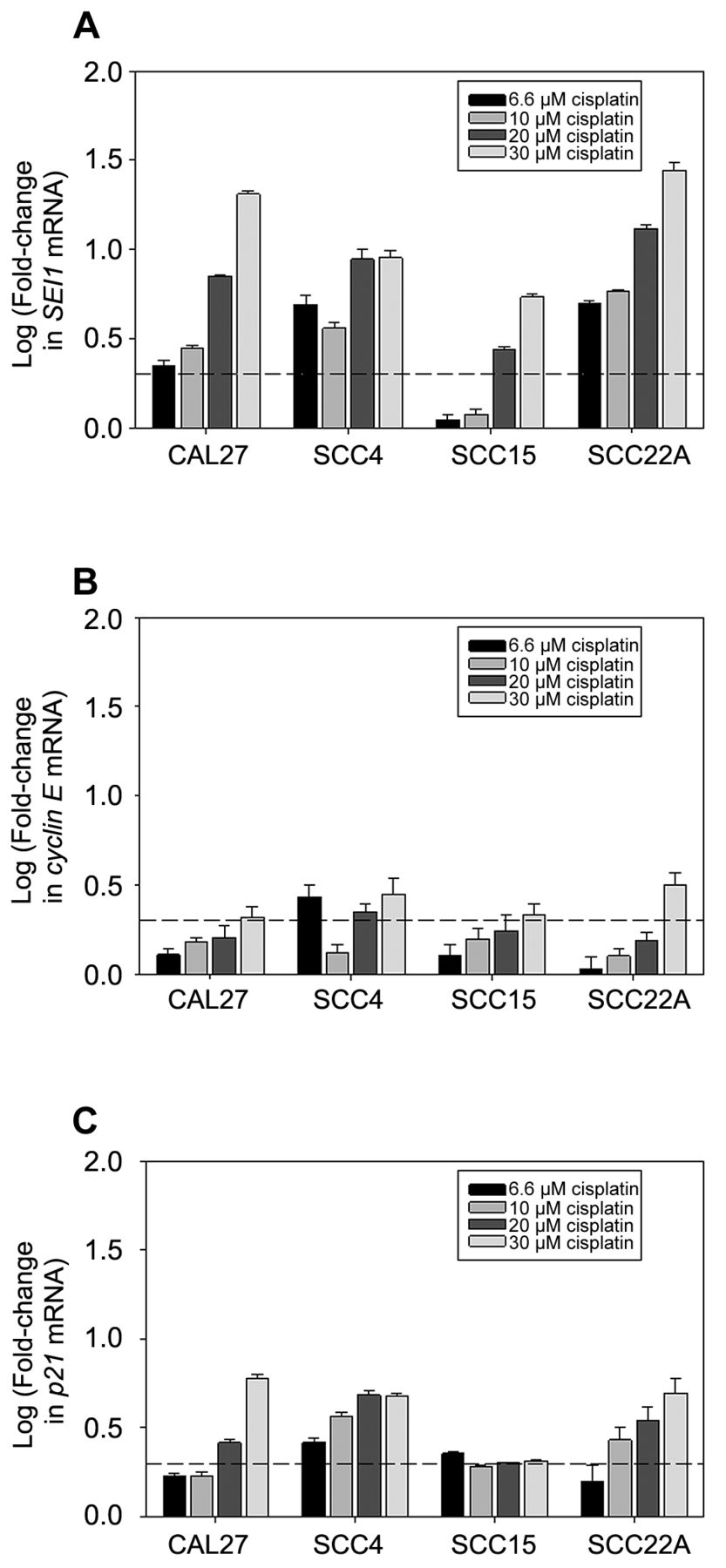
Cisplatin induced the expression of SEI1 (A), cyclin E (B), and p21 (C) in OSCC cells. Pre-validated qRT-PCR-based assays were used to evaluate the mRNA expression levels of target genes. While HPRT1 was used as an endogenous reference for gene expression, cells without cisplatin treatment were used as controls. Change in mRNA expression of a target gene was defined using the 2−ΔΔCq method. Cisplatin treatment experiments were performed in triplicate. Each expression quantitation assay was repeated at least three times. Data are presented as mean±standard deviation. The dash line over the horizontal axis represents a 2-fold increase in gene expression.

Impacts of cisplatin on the expression of CDK4 (A), gankyrin (B), and cyclin D1 (C) in OSCC cells. Legends are similar as in Figure 2. While the dash line over the horizontal axis represents a 2-fold increase in gene expression, the dash line below the horizontal axis in C indicates a 2-fold decrease in gene expression.

Impacts of 5FU on the expression of SEI1 (A), cyclin E (B), and p21 (C) in OSCC cells. Legends are similar as in Figure 2. While the dash line over the horizontal axis represents a 2-fold increase in gene expression, the dash line below the horizontal axis indicates a 2-fold decrease in gene expression.
Discussion
Despite significant efforts to develop novel therapeutics, chemotherapy remains to be a crucial part of the standard care of cancer patients. However, resistance to chemotherapeutic agents is becoming a big challenge in cancer patient care. Recent studies have indicated that p34SEI1, an oncogenic protein functioning in cell cycle progression, apoptosis, and chromosomal stability (10-18), might play important roles in the development of resistance to cisplatin, one of the most commonly used chemotherapeutic agents in cancer patient care (21-24). Frequently overexpressed in a number of human cancers and cancer cell lines (12, 15, 20), p34SEI1 has been found to be able to increase the survival of various types of tumor cells through 1) promoting cell cycle progression (10-12) and 2) inhibiting DNA damage repair and apoptosis (13-16). While overexpression of SEI1 has been regarded as a prognostic biomarker for poor overall survival in patients with liver, ovarian, and gastric cancer (21), recent studies have shown that DNA damage agents, such as radiation and cisplatin, significantly induce the expression of SEI1 in different cancer cells, and such up-regulation inhibits DNA damage repair and apoptosis, thus enabling cells to escape from cell death (22-24). In our current study, the expression of SEI1 in all tested OSCC cell lines was significantly up-regulated upon cisplatin treatment, and such up-regulation appeared to be specific to a certain extent. On one hand, among a selected group of oncogenes that are frequently overexpressed in OSCC in a coordinate manner with SEI1 (21), cisplatin-induced up-regulation was only observed in SEI1 and SEI1-modulated genes (namely, cyclin E and p21); on the other hand, up-regulation of SEI1 was associated with cisplatin, not 5FU.
While molecular mechanisms underlying cisplatin-induced up-regulation of SEI1 remain to be further elucidated, it is likely that a negative feedback loop exists in cancer cells. Upon cisplatin treatment, cisplatin-induced DNA damage accumulates in cells and tends to drive these cells into apoptosis; such DNA damage accumulation in turn activates the transcription of SEI1, which consequently inhibits DNA damage-induced apoptosis and enables these cells counteract against cisplatin-induced cell death and survive. From this perspective, aberrant SEI1 expression is not only a potential prognostic factor for cancer survival but also a factor associated with resistance to cisplatin (DNA damage agents in general). As such, p34SEI1 represents a novel chemotherapeutic target in human cancer treatment (33). Small molecules down-regulating SEI1 at the transcription or protein level may have potential as monotherapy agents or in combination with radiation and/or chemotherapeutic agents such as cisplatin. These molecules may potentiate cisplatin, as well as radiation and other DNA damage agents, in cancer therapy by inhibiting the development of resistance.
Footnotes
Authors' Contributions
JL and MP designed the experiments, analysed the data, and wrote the manuscript. JL and ZV performed the experiments. All the Authors read and approved the final manuscript.
This article is freely accessible online.
Conflicts of Interest
The Authors declare no conflict of interest.
- Received November 21, 2019.
- Revision received November 26, 2019.
- Accepted November 27, 2019.
- Copyright© 2020, International Institute of Anticancer Research (Dr. George J. Delinasios), All rights reserved
References
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.