


Abstract
Overexpression of acyl-coenzyme A:cholesterol acyltransferase (ACAT) results in increased cholesteryl ester levels and has been involved in a variety of cancer types. As a consequence, cholesterol metabolism has raised interest as a potential target for cancer treatment. Inhibition of ACAT results in suppression of proliferation in a range of cancer cell types both in vitro and in vivo. The exact mechanism of this phenomenon is being investigated, and the most important findings are presented in this review.
General cholesterol metabolism. Cholesterol is an amphipathic lipid that is an important element of cell membranes and the outer layer of lipoproteins. The presence of cholesterol in cell membranes limits the permeability and fluidity of these structures, which is necessary to sustain their integrity. Moreover, the cholesterol content in membranes determines the affinity for particular proteins, consequently influencing the biochemical functions of the membrane (1, 2). Cholesterol in the cell membrane is not evenly distributed, and regions where the concentration of cholesterol is increased are called lipid rafts (3). In tissues and in plasma, cholesterol occurs in a free form or is bound to a long chain fatty acids (most commonly oleic acid), forming cholesteryl esters (CE) (4). In plasma, both forms are transported in lipoproteins. In cells, free cholesterol can be found in cell membranes, whereas cholesteryl esters are stored in lipid droplets in the cytosol. Esterified cholesterol is a spare form of cholesterol and provides the possibility of filling free cholesterol shortages in cells. Cholesterol is not just a component of cell membranes. In steroidogenic tissues, this lipid is a precursor of steroid hormones (glucocorticoids, mineralocorticoids and sex hormones). In skin tissue, vitamin D3 is synthetized from 7-dehydrocholesterol in the presence of UV radiation. In the liver, cholesterol is converted into bile acids, which participate in lipid digestion. Excess cholesterol can be removed from an organism in the form of bile (Figure 1). Additionally, dissolution of cholesterol in bile protects against gallstone formation (5). Recently, studies have investigated the impact of cholesterol metabolism on the immune response (6).
In humans, approximately ¾ of the cholesterol comes from de novo synthesis, while the remaining amount is acquired from diet. Almost all tissues that contain nucleated cells are capable of cholesterol synthesis (7). This process occurs in the endoplasmic reticulum, the cytosol and in peroxisomes (8). Cholesterol is synthesized from two-carbon units. The basic building block for cholesterol synthesis is acetyl-CoA. In mammalian cells, acetyl-CoA is mostly synthesized in mitochondria through two processes: i) oxidative decarboxylation of pyruvate and ii) fatty acid oxidation (acyl-CoA β-oxidation). A primary source of pyruvate for this reaction is glucose, but pyruvate can also be formed either from lactate or alanine or indirectly from other glycogenic amino acids. Smaller amounts of acetyl-CoA can also be formed as a result of other amino acid metabolism. A key point in the regulation of cholesterol synthesis from acetyl-CoA, is the synthesis of mevalonate from HMG-CoA, which is catalyzed by 3-hydroxy-3-methylglutaryl-CoA reductase (HMG-CoA reductase; HMGCR) (9). To a great extent, the rate of synthesis and the level of cholesterol depend on the activity of this enzyme and the availability of the substrate (acetyl-CoA). Another source of cholesterol for cells is VLDL (very low density lipoprotein) metabolism. VLDLs are synthesized and subsequently released into the blood by hepatocytes. Their subsequent metabolism leads to LDL (low density lipoprotein) formation. Both free and esterified cholesterol can be collected by cells through LDL phagocytosis with the participation of cell membrane-bound LDL receptors (10).
For most types of cells, the level of free, non-esterified cholesterol – as a component of cell membranes – is of key importance. Cells strictly regulate their amount of free cholesterol. Overly high or low levels of free cholesterol in the cell membrane can drastically change the physicochemical properties of the membrane, leading to impairment of its function. It is considered that a high level of free cholesterol is toxic. Therefore, cells have developed systems to eliminate excess cholesterol. Cholesterol efflux occurs through ABCA1/G1 receptors with the participation of plasma high density lipoproteins (HDLs). In HDLs, cholesterol is esterified by LCAT (lecithin-cholesterol acyltransferase) and subsequently transferred to the liver in a process known as reverse cholesterol transport. Another way that cells eliminate excess cholesterol is the synthesis and subsequent release of oxysterols—hydroxylated forms of cholesterol (e.g., 27-hydroxycholesterol, 24S-hydroxycholesterol, 4β-hydroxycholesterol, and 7α-hydroxycholesterol) into the blood. This process is particularly characteristic of excess cholesterol elimination in the brain (11). Finally, a very fast way to lower the free cholesterol pool is esterification catalyzed by acyl-coenzyme A:cholesterol acyltransferase (ACAT). This reaction allows excess cholesterol to be stored as cholesteryl esters, which can be relatively quickly released by cholesteryl ester hydrolase (12). Considering that free cholesterol is crucial for cell growth and function, investigating ACAT activity regulation in various cells, including cancer cells, may be meaningful and clinically important.
ACAT structure and mechanism of action. Acyl-coenzyme A:cholesterol acyltransferase is one of the key enzymes involved with cellular cholesterol homeostasis. In mammals, two isoforms of this enzyme, encoded by two separate genes, are known: i) ACAT1 and ii) ACAT2. ACAT1 (50 kDa in humans) is the major isoenzyme expressed in all examined tissues, while ACAT2 (46 kDa in humans) is mainly localized in enterocytes and hepatocytes (13). The fact that ACAT2 is found only in lipoprotein-secreting cells of both the liver and intestine emphasizes the possibility that this enzyme may interact in some way with the lipoprotein particle assembly machinery to facilitate lipoprotein particle formation. The differential pattern of distribution supports the hypothesis that in contrast to ACAT2, ACAT1 is involved in intracellular cholesterol homeostasis (14).
ACATs are localized in the ER and belong to the membrane-bound O-acyltransferase (MBOAT) enzyme family, which comprises 11 members, including acyl-CoA:diacylglycerol acyltransferase 1 (DGAT1), lysophosphatidic acyltransferases (LPATs) and ghrelin octanoyl-coenzyme A acyltransferase (15). MBOATs are membrane enzymes that use long-chain or medium-chain acyl-CoA and hydrophobic substances as substrates. The reaction mechanism involves two important active sites: i) a histidine residue located within a long stretch of hydrophobic residues and ii) an asparagine residue within the hydrophilic peptide region.
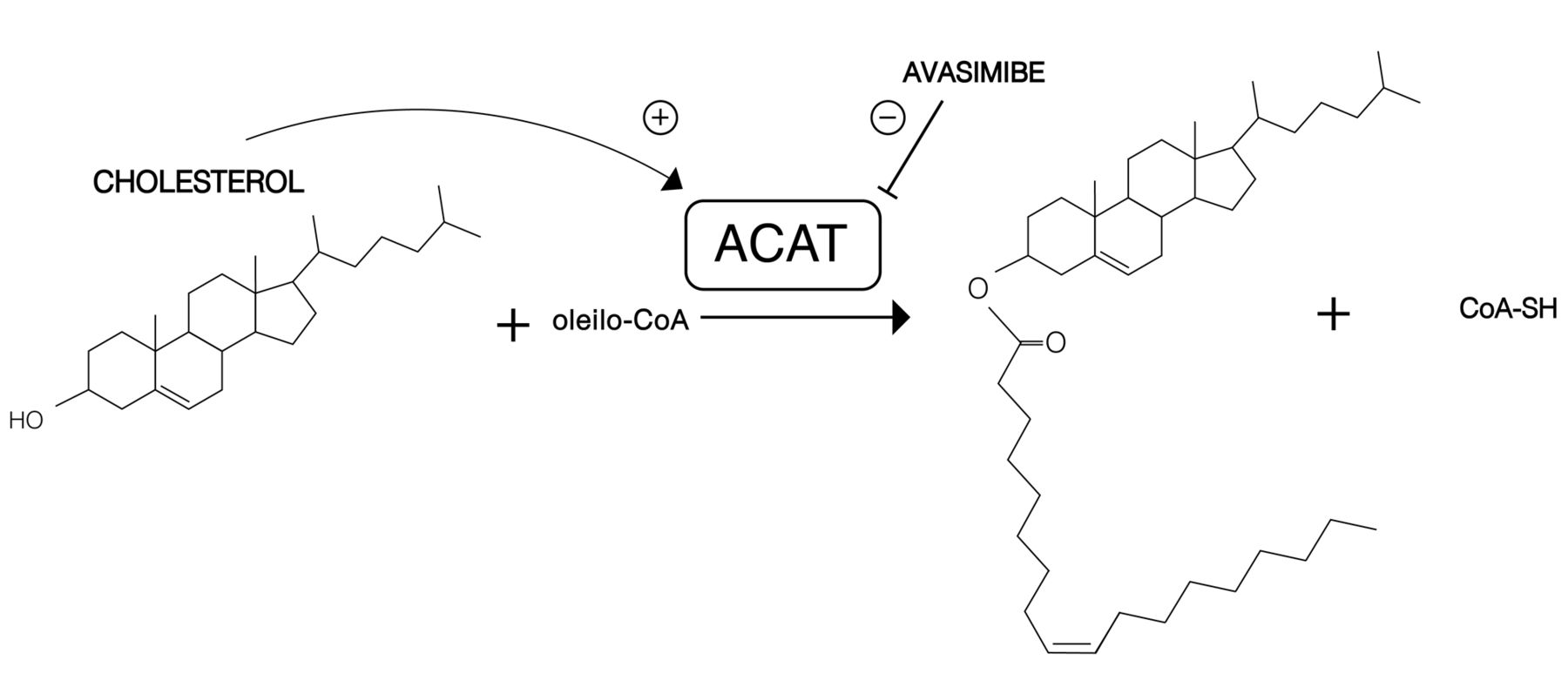
The main reaction catalyzed by ACATs is the conversion of cholesterol to cholesteryl esters (Figure 2). Among long-chain fatty acids, oleic acid (18:1) is the preferred substrate for ACAT1, whereas ACAT2 esterifies i) 18:1, ii) eicosapentaenoic acid (20:5) and iii) docosapentaenoic acid (22:5) fatty acids equally (16). Furthermore, ACATs are known as sterol O-acyltransferases (SOATs) because both ACAT1 and ACAT2 can use a variety of sterols (containing a 3β hydroxyl group in steroid ring A) as substrates. It is worth noting that sterols not only are substrates in reactions catalyzed by ACATs but are also activators of these enzymes (17, 18). It has been shown that cholesterol is both the best substrate and the best activator among the tested substrates (oxysterols, pregnenolone, DHEA, plant and yeast sterols, some synthetic sterol analogs) (19, 20). A study by Liu et al., has shown that epicholesterol, which contains a 3-alpha-OH group in steroid ring A, is neither a substrate nor an activator of this enzyme (21). However, it was shown that epicholesterol induced positive changes in the ACAT1 fluorescence spectrum. Although the magnitude of these spectral changes was large, the changes induced by cholesterol are much larger (22). It has been demonstrated that the addition of cholesterol to the assay mixture increases the rate of pregnenolone esterification (catalyzed by both ACAT1 and ACAT2) by 100-fold (20). Based on their research on pregnenolone and cholesterol esterification, Rogers et al., have proposed a working model to explain ACAT1 allosterism. ACAT1 is homotetrameric but may act as a double dimer. Each dimer may contain two identical sterol substrate sites (site S) and one or two sterol activator sites (site A). Site S can bind a variety of sterols containing a 3-beta-hydroxyl group in steroid ring A but preferentially binds pregnenolone. It is interesting that site A cannot bind pregnenolone, however, among many other tested sterols, it preferentially binds cholesterol. When only pregnenolone is present, the esterification reaction proceeds at a very low rate because pregnenolone (in the absence of cholesterol) fails to trigger appropriate conformational changes in the enzyme. Cholesterol may effectively bind to both site A and site S. The binding of cholesterol at site A causes conformational changes, enabling the enzyme to bind its substrates (pregnenolone and cholesterol) tighter to site S and to catalyze esterification much more efficiently (20).
ACAT in cancer. Lipid metabolism plays an important role in cancer development and in cell migration, invasion and metastasis. The activation of fatty acid synthesis is required for carcinogenesis and for tumor cell survival. Overexpression of genes encoding lipogenic enzymes (involved in de novo lipogenesis) has been shown in many human cancer types (23). It is now known that cholesterol metabolism plays a substantial role in tumorigenesis. Because ACAT1 is involved in intracellular cholesterol homeostasis, possibly through the maintenance of the appropriate level of esterified cholesterol within cells to support membrane stability, this enzyme became the target of research in tumor cells.

Simplified scheme of cholesterol metabolism.
Overexpression of ACAT1 followed by increased cholesteryl esters accumulation in lipid droplets (LDs) has been shown in a variety of cancer types. Cancer cells preferentially form CE for storage and reuse if needed. It has also been shown that breast cancer cell lines (MDA-MB-436 and MDA-MB-231) contain a greater number of cytoplasmic lipid droplets than luminal MCF-7 cells and that ACAT1 inhibition by the inhibitor CP-113818 reduces the proliferation of breast cancer cells (24). Mulas et al., have revealed that leukemic cells have increased levels of cholesteryl esters and reduced levels of free cholesterol (25). According to Yue et al. 2014, CE accumulation is a consequence of the loss of the tumor suppressor PTEN (phosphatase and tensin homolog), and of the subsequent activation of the PI3K/AKT pathway (26). Moreover, these authors suggest a novel quantitative approach for classifying prostate cancers based on cholesteryl ester accumulation. This study has also shown that CE storage potentiates PI3K-dependent SREBP activity, thereby fueling cancer aggressiveness. A correlation between ACAT1 overexpression and patient prognosis was later confirmed in pancreatic cancer (27).

The main reaction catalyzed by ACATs. Cholesterol is a substrate as well as a activator in this reaction. Based on Rogers et al. (17).
Further investigations have shown that CE depletion caused by pharmacological inhibition of ACAT results in the suppression of cancer cell proliferation, migration and invasion in various cancer cell lines as well as in reduction of tumor growth in vivo. Exposure of leukemic cells to inhibitors of cholesterol esterification, such as progesterone and SaH–58035, can cause a 60% reduction in their proliferation rate (25). Subsequent studies have shown that another ACAT inhibitor, avasimibe, inhibits the growth of glioma cell lines (U87, A172 and GL261) by inducing cell cycle arrest and apoptosis (28). A similar mechanism has been observed in pancreatic cancer, where Li et al., have shown a high level of CE in LDs and overexpression of ACAT-1 in human pancreatic cancer cells and cell lines (MIA PaCa-2 and PANC-1) (29). Inhibition of ACAT by avasimibe effectively blocked CE accumulation in those cell lines. Another study has shown that avasimibe reduces the proliferative and invasive capabilities of cells as well as tumor growth in mouse models of high-grade prostate cancer (26). Similar effects have been observed in colon cancer, pancreas and in prostate models (27, 29-31). Moreover, systemic treatment with avasimin (the encapsulated form of avasimibe) notably suppressed tumor growth in mice and extended their survival time (14). ACAT1 has been associated with the function of TLR4 (Toll-like receptor 4) in colorectal cancer progression. Ye et al., have shown that inhibition of TRL4 suppress cell proliferation, migration and invasion. This effect was efficiently abolished when ACAT1 was overexpressed, providing evidence that the antitumoral effect of TRL4 suppression is mediated by ACAT1 (30). Lee et al., have observed a reduction in metastatic lesion growth in a prostate cancer mouse model and have suggested that CE depletion via ACAT inhibition reduces the metastatic potential through the upregulation of multiple regulators that negatively impact metastasis, especially by downregulating the Wnt/β-catenin pathway, a vital pathway involved in metastasis, and by blocking Wnt3a secretion through a reduction in monounsaturated fatty acid levels, which limits Wnt3a acylation (31) .
The most recent studies have concentrated on the potential of avasimibe in combination cancer therapy. Bandyopadhyay et al., have shown suppressed proliferation in chronic myelogenous leukemia (CML) (in K562R cells rendered imatinib-resistant and in a K562R xenograft model) (32). According to the authors, the synergistic effect of avasimibe and the chemotherapeutic drug imatinib may be due to downregulation of the MAPK pathway by avasimibe, which sensitizes CML cells (primary cells from a patient with BCR-ABL mutation-independent imatinib resistance) to imatinib treatment. Other researchers have revealed similar effects of combination therapy. Li et al., have found that the combination of avasimibe and the chemotherapy drug gemcitabine reduce cell viability and tumor growth in pancreatic cancer (PDAC; PaCa-2 and G3K cells). The authors have shown that avasimibe overcomes gemcitabine resistance by downregulating the protein kinase B (Akt) signaling pathway, suggesting that dysregulation of cholesterol homeostasis inactivates Akt signaling possibly through a feedback mechanism and re-sensitizes pancreatic cancer cells to gemcitabine treatment (29).
These data suggest that the level of ACAT expression is a potential prognostic marker for some types of cancer. ACAT inhibition has been suggested to suppress cancer cell proliferation (Table I). Taken together, these results suggest that pathways controlling cholesterol esterification might represent promising targets for novel anticancer strategies.
Experimental studies on ACAT inhibition in cancer treatment.
Potential mechanisms of cholesterol cytotoxicity in cancer cells. As described above, several models indicate that ACAT inhibition causes cytotoxicity. In general, inhibition of ACAT leads to an increase in intracellular free cholesterol, which is then incorporated into the cell membrane. Enrichment of cell membranes in cholesterol leads to changes in membrane proteins involved in intracellular signaling activity, which may be a cause of signaling dysregulation; the result is, thus, toxic and can lead to loss of cell viability (33-35).
The key regulator of cholesterol metabolism is SREBP-2 (sterol regulatory element binding protein 2), a transcription factor anchored to the ER membrane (36). The activity of this protein as a transcription factor is mainly regulated by the levels of free cholesterol in the ER membrane. Several regulatory proteins associated with SREBP2 participate in this regulation. These proteins include SCAP (SREBP cleavage-activating protein), INSING (insulin-induced gene) and ERLINS (ER lipid raft-associated protein). All of these proteins have sterol-sensitive domains. Cholesterol binds to SCAP and ERLINS, while oxysterols bind to INSING (37). When cholesterol levels in the ER are low, SCAP transfers SREBP2 to the Golgi apparatus for proteolytic activation, which releases a soluble transcription factor (the active form). The active form of SREBP enters the nucleus and activates genes involved in cholesterol synthesis and uptake (e.g., the LDL receptor gene or HMG-CoA reductase gene). When cellular cholesterol levels are sufficient or excessive, INSING, supported by ERLINS, retains the SCAP/SREBP complex in the ER. In prostate cancer cells, SREBP-2 activity is induced by hyperactivation of PI3K/AKT/mTOR signaling pathways caused by PTEN loss (38). This hyperactivation leads to an increase in the expression of the LDL receptor and, consequently, an increase in the cellular uptake of lipids, including essential (unsaturated) fatty acids (FAs). Unsaturated FAs (mainly arachidonic acid) are important precursors for signaling lipids, such as prostaglandins, which regulate the proliferation, migration and invasion of cancer cells (39). Inhibition of ACAT, with elevated levels of free cholesterol in the ER membrane, leads to the suppression of SREBP-2 precursor transport from the ER to the Golgi. Inhibition of SREBP-2 activity causes changes in the regulation of gene expression, such as decreasing the transcription of the LDL-R gene, and in lipid uptake by cells. In summary, ACAT-1 inhibition disrupts cholesterol esterification and results in accumulation of non-esterified cholesterol, inhibition of SREBP activity, a reduction in lipid uptake, and inhibition of growth (38, 40, 41).
The cytotoxicity of cholesterol may also be associated with the regulation of caveolin 1 gene expression by SREBP2. Caveolins are a family of 22-kDa proteins implicated in cholesterol homeostasis, signal transduction, and vesicle trafficking (42). Caveolin-1 not only is an essential caveolar scaffolding protein but it also acts as a tumor modulator involved in cellular transformation, tumor growth and cell metastasis. Upregulation of the caveolin-1 gene has been demonstrated to be associated with the promotion of cell proliferation and invasion in the same cancers (e.g., bladder, esophageal and prostate cancer and thyroid papillary carcinoma) (43, 44). Therefore, it is possible that inactivation of SREBP2 leads to down-regulation of the caveolin-1 gene. Since caveolin participates in tumorigenesis through the caveolin-1/MAPK pathway, caveolin deficiency leads to the suppression of this pathway and, consequently, a reduction in cancer aggressiveness (27, 45).
Another possible mechanism for cancer cells apoptosis is the induction of ER stress by elevated free cholesterol levels in cancer cells. Warner et al., have shown that an increase in the free cholesterol level leads to ER stress and, consequently, to apoptosis (46). In colon cancer cells treated with an ACAT inhibitor, significantly increased levels of ER stress markers, such as GRP78 (78-kDa glucose-regulated protein), ATF4 (activating transcription factor 4) and CHOP (C/EBP homologous protein), have been observed (27) .
In turn, Lee et al., have noted a pathway for a reduction in tumor expansion associated with cholesterol metabolism (31). According to their findings, depletion of CE limits Wnt3a acylation and, consequently, its secretion. Wnt proteins are secreted, lipid-modified proteins that regulate embryonic development and tissue renewal in adults. Wnt acylation is also required for the binding of Wnt to its cell surface receptor and subsequent Wnt signaling activity (47). Since Wnt is a part of the Wnt/β-catenin pathway, a pathway vital for metastasis, its suppression inhibits the growth of metastatic lesions and the migration of cancer cells (48).
It is also worth noting that elevated free cholesterol levels in cell membranes inhibit HMGCR activity. This enzyme is located in the ER membrane and possesses a sterol-sensing domain. An increase in the free cholesterol concentration in the ER membrane leads to enzymatic degradation of HMGCR in the proteasome (9, 49). Consequently, de novo synthesis not only of cholesterol but also of nonsterol products (prenyl groups), whose synthesis pathway partially overlaps with that of cholesterol, is inhibited. Prenyl groups attached to proteins are derived from 5-carbon intermediates in the cholesterol biosynthetic pathway to form farnesyl (15 carbons) and geranylgeranyl (20 carbons) groups. Protein prenylation is an essential eukaryotic cell protein modification required for the membrane anchoring of many regulatory proteins, such as oncogene RAS, in cancer cells (50).
Conclusion
Recent studies indicate that cholesterol metabolism is highly correlated not only with cardiovascular diseases, but also with tumorigenesis. Therefore, controlling cholesterol esterification may be a new approach to inhibit cancer cell proliferation and metastasis formation. The relationship between the level of CE accumulation in LDs and the grade of cancer aggressiveness is well documented; however, the mechanism of this phenomenon has not been precisely explained to date.
Acknowledgments
This work was supported by the National Science Centre of Poland (grant no. 2016/22/E/NZ4/00665) and Medical University of Gdansk (grants no. ST-40, and ST-41).
Footnotes
Authors' Contributions
JZ and ES studied the literature and wrote the manuscript and TS revised the manuscript. All authors read and approved the manuscript.
Conflicts of Interest
The Authors declare that they have no conflicts of interest.
- Received May 15, 2019.
- Revision received June 19, 2019.
- Accepted June 20, 2019.
- Copyright© 2019, International Institute of Anticancer Research (Dr. George J. Delinasios), All rights reserved
References
In this issue





{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.