


Abstract
Background/Aim: Chemotherapy is an important first-line treatment for oesophageal squamous cell carcinoma (ESCC). However, there are few secondary options. Olaparib, a poly (ADP-ribose) polymerase (PARP) inhibitor, enhances the cytotoxicity of various anticancer drugs and has been used to treat advanced ovarian and breast cancers. This study examined the effect of olaparib on the cytotoxicity of anticancer drugs in ESCC cell lines. Materials and Methods: ESCC KYSE70 and KYSE140 cells were grown in Dulbecco's modified Eagle's medium and treated with 5-fluorouracil (5-FU), cisplatin, docetaxel, doxorubicin, SN-38, or temozolomide without or with olaparib. Results: Olaparib enhanced the cytotoxicity of all tested anticancer drugs and increased the effects of cisplatin, doxorubicin, SN-38, and temozolomide synergistically. These anticancer drugs caused the accumulation of phospho-histone H2AX Ser139 (γH2AX), a biomarker of DNA damage, and olaparib increased this accumulation. Conclusion: PARP inhibitors may potentiate the anticancer activity of DNA-damaging agents in ESCC patients synergistically.
Oesophageal squamous cell carcinoma (ESCC) is one of the most lethal malignant diseases worldwide and is the sixth leading cause of cancer-related deaths (1). The survival rate of ESCC patients is low (10-30%) in most countries, although that of colorectal cancer patients is over 70% (2). The poor outcome of ESCC may result from the difficulty of early detection because its subjective symptoms are not noticeable in the early stage of the disease. Furthermore, ESCC patients are more likely to have tumours that invade and metastasise to other organs (3). Hence, it is important to initiate treatment against ESCC patients with anticancer drugs as early as possible to improve the prognosis.
5-Fluorouracil (5-FU), cisplatin (CDDP), and docetaxel are reliable and effective agents as first-line chemotherapy for ESCC, achieving response rates of 30-70% (4-6). However, 30% or more patients who receive first-line chemotherapy fail to respond to treatment (4-6), and there is no consensus on second-line chemotherapy for ESCC (7). Therefore, it is necessary to establish an effective second-line chemotherapy for ESCC.
Poly (ADP-ribose) polymerase (PARP) inhibitors are novel molecular-targeted drugs for cancer therapy that inhibit PARP1 enzymatic activity. The PARP inhibitor, olaparib, has been used in Europe, the USA, and Japan for treating advanced ovarian and breast cancers. Especially, olaparib is more effective for patients who have breast cancer susceptibility gene (BRCA) 1/2 mutations in the tumour (8, 9).
PARP1 adds one or more ADP-ribose units to itself or some target proteins, and modulates the DNA repair pathways or transcription factors (10). Therefore, PARP inhibitors are not only effective molecular-targeted drugs for treatment when used alone, but also sensitize tumours to other anticancer drugs (10). For example, co-administration of olaparib improved the prognosis of ovarian and breast cancer patients undergoing treatment with carboplatin and paclitaxel in clinical studies (11), and the combination of a PARP inhibitor and irinotecan was effective against human colorectal cancer cells in a xenograft mouse model (12). In ESCC cell lines, olaparib enhanced the cytotoxicity of CDDP synergistically (13). Therefore, it is possible that olaparib can improve the therapeutic effect of anticancer drugs in ESCC. However, the types of anticancer drugs that can be combined with olaparib to enhance the efficacy against ESCC are not known.
The purpose of this study was to clarify the types of anticancer drugs that can interact synergistically with olaparib. To achieve this purpose, the effects of olaparib on the cytotoxicity of various anticancer drugs in ESCC cell lines were investigated.
Materials and Methods
Chemicals and reagents. Olaparib was obtained from LKT Laboratories (St. Paul, MN, USA). 5-FU was purchased from Sigma-Aldrich (St. Louis, MO, USA). CDDP, doxorubicin, and the DNA-dependent protein kinase (DNA-PK) inhibitor, NU7441, were purchased from Wako Pure Chemical Industries (Osaka, Japan). Docetaxel, 7-ethyl-10-hydroxy-camptothecin (SN-38), and temozolomide were purchased from Tokyo Chemical Industry (Tokyo, Japan).
Cell culture. The ESCC KYSE70 and KYSE140 cell lines were obtained from the Health Science Research Resources Bank (Osaka, Japan) (14). Cells were maintained in Dulbecco's modified Eagle's medium (Life Technologies, Grand Island, NY, USA) supplemented with 10% heat-inactivated foetal bovine serum (GE Healthcare, Little Chalfont, UK), 100 units/ml of penicillin, and 100 mg/ml of streptomycin (Nacalai Tesque, Kyoto, Japan). Cells were grown in an atmosphere of 5% CO2 and 95% air at 37°C.
Growth inhibition assay. Cell growth was evaluated using the CellQuanti-Blue Cell Viability Assay Kit (BioAssay Systems, Hayward, CA, USA) as described previously (15). In brief, cells (1×103) were seeded onto a 96-well plate in 100 μl of medium per well. After 24 h, cells were exposed continuously for seven days to anticancer drugs with or without olaparib (5 μM with KYSE70 and 1 μM with KYSE140 cells) and NU7441 (0.5 μM with KYSE70 cells). The medium was then replaced with fresh medium containing CellQuanti-Blue reagent solution (1:10). After 5 h, the fluorescence intensity was measured by a GENios microplate reader (Tecan, Seestrasse, Switzerland) at excitation and emission wavelengths of 535 and 590 nm, respectively. Half-maximal inhibitory concentrations (IC50s) were calculated according to the sigmoid inhibitory effect model (eq. 1) using the nonlinear least-squares fitting method (Solver, Microsoft Excel).

E and Emax represent the surviving fraction (% of control) and its maximum, respectively. C and γ are the drug concentration in the medium and the sigmoidicity factor, respectively (16).
Clonogenicity assay. KYSE70 cells (1×103) were seeded onto six-well plates in 2 ml of medium per well. After 24 h, cells were exposed continuously for two weeks to the anticancer drugs with or without olaparib (2.5 μM). Cell colonies were fixed and stained with a 60% methanol solution containing 0.1 w/v% methylene blue. Colony formation was analysed semi-quantitatively by determining the dyed cell colonies area using Image J software (National Institutes of Health, Bethesda, MD, USA) as described previously (17). Interactions between anticancer drug and olaparib were evaluated using the expected value (EXP) (eq. 2). Synergism was determined when the value was less than EXP, additivity when the value was the same as EXP, and antagonism when the value was more than EXP (18).

Western blot analysis. Western blot analysis was performed using methods described previously (15). KYSE70 cells (2×106) were seeded onto a 60-mm dish. After 47 h, cells were treated with olaparib (10 μM) for 1 h. Then, the cells were treated with anticancer drugs with or without olaparib (10 μM). After 24 h, cells were lysed with CelLytic M (Sigma-Aldrich) containing 1 mM phenylmethylsulfonyl fluoride. Protein concentrations were measured by the Bradford method (19). Proteins (20 μg) were separated using 7.5% (for poly ADP-ribosylated (PAR) proteins, an indicator of PARP activity) or 10% (for β-actin, the reference protein) sodium dodecyl sulphate polyacrylamide gels (SuperSep Ace, Wako) and transferred to polyvinylidene difluoride membranes (ClearTrans SP, Wako). Membranes were blocked in phosphate-buffered saline/0.1% Tween 20 (PBS-T) containing 1% skim milk (Wako). The membranes were incubated at 4°C overnight with anti-PAR mouse monoclonal IgG3κ (1:1000; Trevigen, Gaithersburg, MD, USA) or anti-β-actin mouse monoclonal IgG1 (1:1000; Wako). The anti-PAR antibody was diluted using Can Get Signal Immunoreaction Enhancer Solution I (Toyobo, Osaka, Japan). The anti-β-actin antibody was diluted using PBS-T. Primary antibodies were probed with horseradish peroxidase-conjugated anti-mouse IgG antibodies (GE Healthcare) for PAR (1:10,000; Can Get Signal Immunoreaction Enhancer Solution I, Toyobo) or β-actin (1:25,000; PBS-T). Proteins were detected by the ImmunoStar LD reagent (Wako) and viewed with the VersaDoc 5000 MP imaging system (Bio-Rad, Hercules, CA, USA).
Immunofluorescence analysis. KYSE70 cells (2×104) were seeded onto black 96-well plates in 100 μl of medium per well and incubated for 24 h. The medium was then replaced with fresh medium containing the anticancer drugs with or without olaparib (5 μM). After 24 h, cells were fixed in 4% formaldehyde containing PBS at 4°C for 15 min, permeabilised with 0.1% polyoxyethylene (10) octylphenyl ether in PBS for 30 min and blocked in PBS-T containing 0.1% bovine serum albumin for 60 min at room temperature. Cells were incubated at 4°C overnight with anti-phospho-histone H2AX Ser139 (γH2AX) mouse monoclonal IgG1 (1:200; Merck Millipore, Billerica, MA, USA). The primary antibody was probed with an anti-mouse IgG (H/L), F(ab')2 Fragment Alexa Fluor 488 Conjugate (1:500; Cell Signaling Technology, Danvers, MA, USA). These antibodies were diluted using PBS-T. Cell nuclei were stained with 0.2 μg/ml 4’,6-diamidino-2-phenylindole in PBS. Cells were visualised using an Operetta High Content imaging microscope (PerkinElmer, Waltham, MA, USA). The mean region intensity of Alexa Fluor 488 fluorescence per cell nucleus was analysed using Harmony software (PerkinElmer). At least 300 cells per experimental point were examined.
Statistical analyses. Data are presented as means±standard error. Differences between two groups were performed using the unpaired Student's t-test. Experiments with three or more groups were assessed using a repeated one-way analysis of variance followed by Tukey's honest significant difference test. All analyses were conducted with two-tailed p-values and considered statistically significant when p<0.05.
Results
Olaparib potentiates the growth-inhibitory effect of anticancer drugs in ESCC cell lines. All tested anticancer drugs inhibited the growth of KYSE70 and KYSE140 cells in a concentration-dependent manner. The growth inhibition curves of these anticancer drugs were shifted to lower concentrations by co-treatment with olaparib (Figure 1). Thus, the IC50 values of all anticancer drugs were decreased by olaparib in KYSE70 and KYSE140 cells. Especially, olaparib remarkably decreased the IC50 values of CDDP, doxorubicin, SN38, and temozolomide; the relative sensitivities were more than 2-fold lower in both ESCC cell lines with olaparib (Table I).
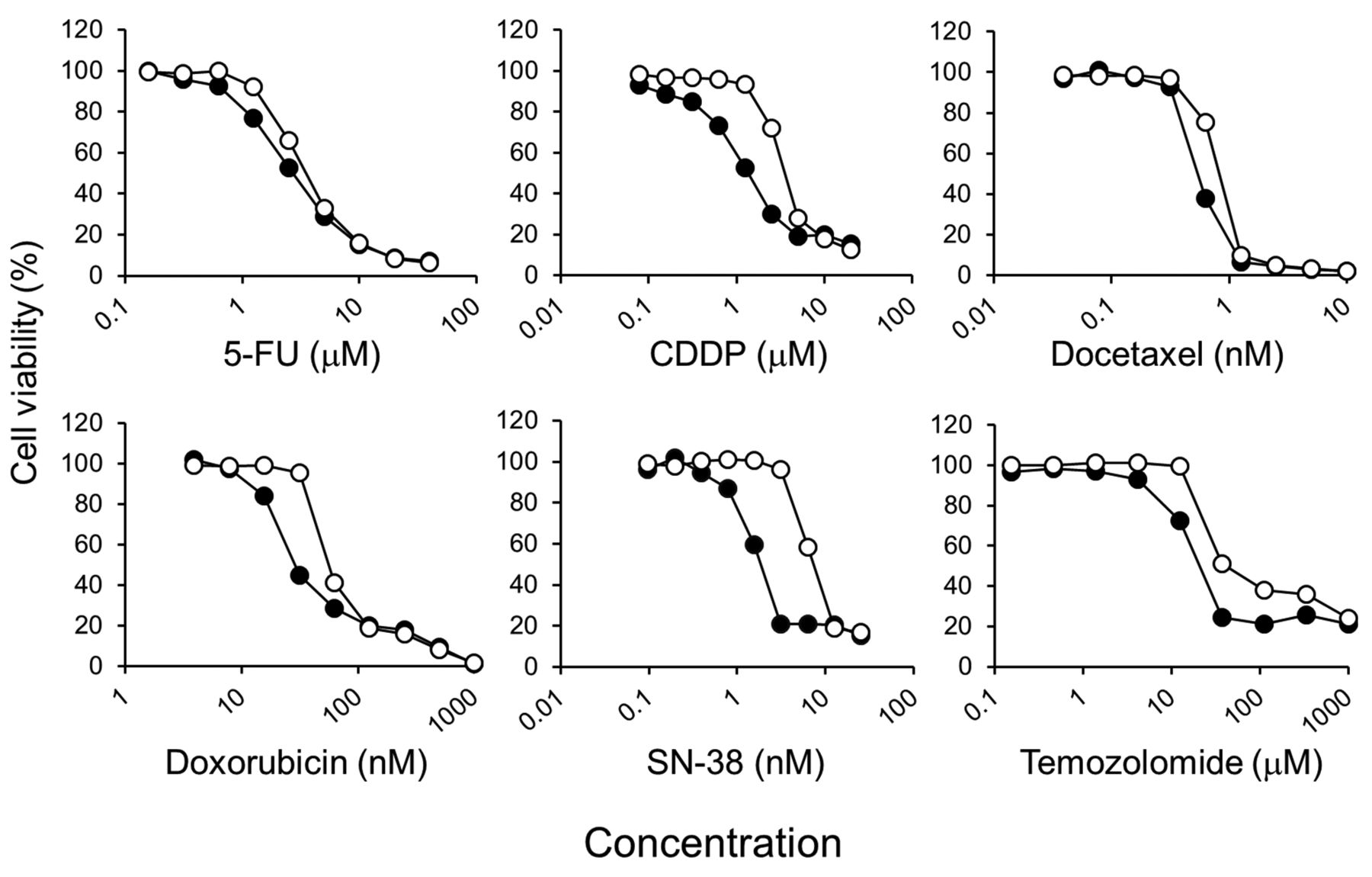
Effects of olaparib on the inhibition of cell growth by anticancer drugs in KYSE70 oesophageal squamous cell carcinoma cells. Cells were seeded onto 96-well plates. After culturing for 24 h, cells were exposed continuously to the indicated drug concentrations for one week without (○) or with olaparib 5 μM (●). Cell viability was determined using the CellQuanti-Blue Cell Viability Assay Kit. Each point represents the mean ± SE (n = 4).
IC50 values of anticancer drugs without or with olaparib in oesophageal squamous cell carcinoma cell lines.
Olaparib decreases the level of PAR proteins in KYSE70 cells. PAR protein expression was observed in KYSE70 cells treated with or without anticancer drugs. Olaparib decreased PAR proteins to nearly undetectable levels in all conditions (Figure 2).

Effect of olaparib on the levels of poly ADP-ribosylated (PAR) proteins in anticancer drug-treated KYSE70 cells. Cells were seeded onto 60-mm dishes. After culturing for 47 h, cells were treated with or without 10 μM olaparib for 1 h, then exposed to the indicated anticancer drugs for 24 h with or without 10 μM olaparib. Total protein was extracted from whole-cell lysates and western blotted for PAR and β-actin (reference) proteins.
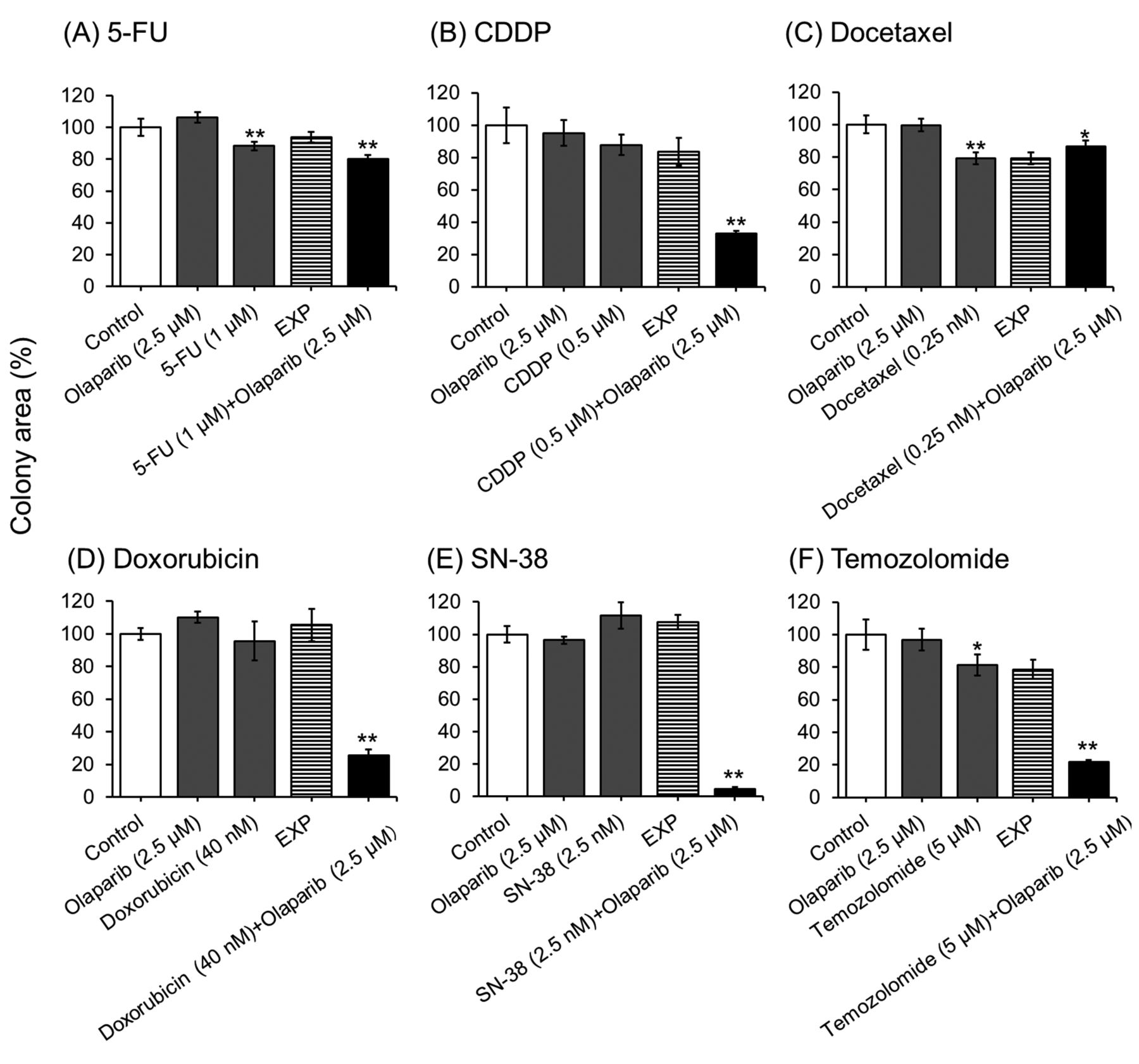
Olaparib synergistically increases the inhibition of cell colony formation by anticancer drugs in KYSE70 cells. 5-FU, docetaxel, and temozolomide alone slightly inhibited the formation of cell colonies. Olaparib, CDDP, doxorubicin, and SN-38 alone did not inhibit the formation of colonies by KYSE70 cells in this experimental condition. However, olaparib markedly enhanced the inhibitory effects of CDDP, doxorubicin, SN-38, and temozolomide on cell colony formation (Figure 3).
Olaparib enhances the accumulation of γH2AX caused by DNA damaging agents in KYSE70 cells. Olaparib significantly increased nuclear γH2AX levels in KYSE70 cells (Figure 4). CDDP, doxorubicin, SN-38, and temozolomide all increased nuclear γH2AX in a concentration-dependent manner. In addition, olaparib additively increased nuclear γH2AX levels in anticancer drug-treated KYSE70 cells (Figure 4).
Effects of the DNA-PK inhibitor, NU7441, on the growth inhibitory activity of anticancer drugs with olaparib in KYSE70 cells. NU7441 potentiated the growth inhibitory effects of doxorubicin plus olaparib, but did not affect that of CDDP, SN-38, or temozolomide plus olaparib in KYSE70 cells (Figure 5).
Discussion
Olaparib enhanced the growth inhibitory effects of anticancer drugs in ESCC cell lines (Figure 1, Table I), and inhibited the expression of PAR proteins in KYSE70 cells treated with anticancer drugs (Figure 2). In addition, olaparib synergistically enhanced the cytotoxic activity of CDDP, doxorubicin, SN-38, and temozolomide against KYSE70 cells (Figure 3). These in vitro results suggest that PARP inhibitors such as olaparib may potentiate the anticancer activity of CDDP, doxorubicin, SN-38, and temozolomide in ESCC patients.
Olaparib synergistically decreased cell colony formation in CDDP-, doxorubicin-, SN-38-, and temozolomide-treated KYSE70 cells, but not in 5-FU- or docetaxel-treated cells (Figure 3). CDDP, doxorubicin, SN-38, and temozolomide increased nuclear γH2AX levels (a marker of DNA double-strand breaks (DSBs)) (20) in KYSE70 cells, but 5-FU and docetaxel did not (Figure 4). Inhibition of PARP1 activity synergistically potentiates the cytotoxicity of DNA damaging agents such as methyl methanesulphonate and hydroxyurea (21, 22). Olaparib and veriparib, another PARP inhibitor, enhance the growth inhibitory effects of 5-fluoro-2’-deoxyuridine, a metabolite of 5-FU, but not 5-FU itself, and cause DNA damage in ovarian cancer cell lines (23). Paclitaxel, a microtubule inhibitor similar to docetaxel, does not induce DNA damage compared to treatment of mouse lymphoma cells with methyl methanesulphonate (24). These results suggest that the effects of anticancer drugs that cause DNA damage are enhanced synergistically by PARP inhibitors in ESCC cell lines.
Olaparib additively increased nuclear γH2AX levels in CDDP-, doxorubicin-, SN-38-, and temozolomide-treated KYSE70 cells (Figure 4). Olaparib treatment increases γH2AX levels in a PARP inhibitor-sensitive breast cancer cell line, but not in PARP inhibitor-resistant cells (25). PARP inhibitors increase DSBs and cell death caused by CDDP (26), doxorubicin (27), SN-38 (28), and temozolomide (29). These results suggest that the synergistic cytotoxicity achieved by combining a PARP inhibitor and DNA damaging agent depends on increasing DSBs in ESCC cell lines.

Effects of olaparib on the inhibition of cell colony formation by anticancer drug-treated KYSE70 cells. Cells were seeded onto 6-well plates. After culturing for 24 h, cells were exposed continuously to the indicated anticancer drugs for 2 weeks with or without 2.5 μM olaparib. Cell colonies were stained with 0.1% methylene blue in 60% methanol. Each bar represents the mean±SE (n=3). Significant differences were determined by an ANOVA followed by Tukey's test (*p<0.05, **p<0.01 vs. control).
NU7441 delays the repair of DNA DSBs caused by topoisomerase II inhibitors, and sensitises cells to the toxicity of doxorubicin and etoposide (30, 31). NU7441 enhanced the growth inhibitory effects of doxorubicin combined with olaparib (Figure 5). Furthermore, inhibiting DNA-PK affected only the sensitivity of doxorubicin in an ESCC cell line, and did not affect the synergism between doxorubicin and olaparib. On the other hand, inhibiting DNA-PK did not affect the growth inhibitory effects of CDDP, SN-38, or temozolomide without or with olaparib in KYSE70 cells (Figure 5).
NU7441 suppresses the chromosomal aberrations and growth inhibitory effects caused by veliparib in a BRCA-mutated ovarian cancer cell line (32). The enzymatic activity of DNA-PK is critical for DNA repair, especially via the classical non-homologous end joining pathway, also known as the error-prone DNA repair pathway (33). NU7441 does not show toxicity in cells with wild-type BRCA. However, BRCA-mutated cells show high sensitivity to NU7441 (34). As shown in the Cancer Cell Line Encyclopedia (https://portals.broadinstitute.org/), KYSE70 cells do not have a BRCA mutation. Hence, these results suggest that inhibition of the classical non-homologous end joining pathway does not affect the synergistic cytotoxicity of olaparib with DNA damaging agents in ESCC cell lines.
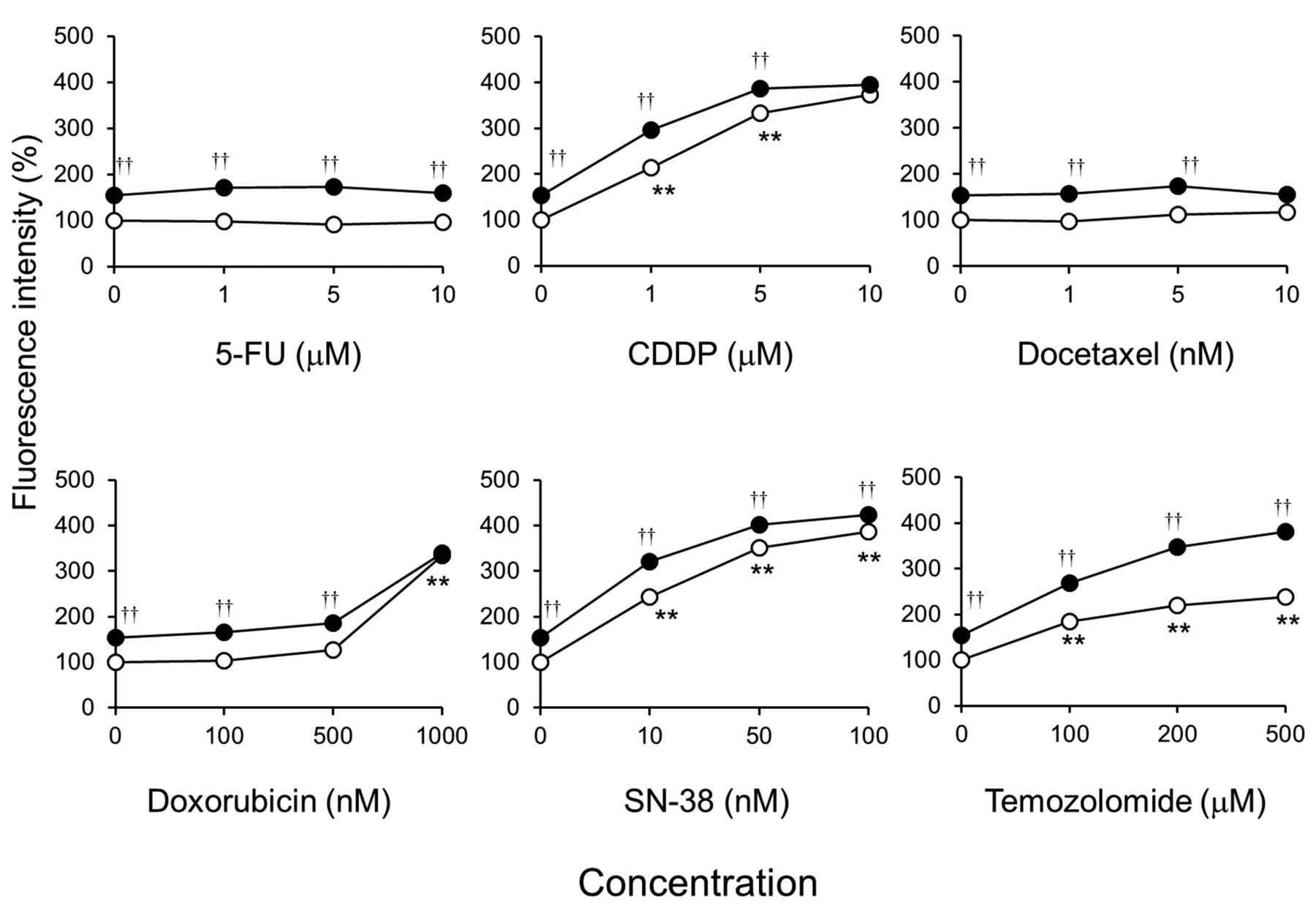
Effects of olaparib on the accumulation of γH2AX caused by anticancer drugs in KYSE70 cells. Cells were seeded onto black 96-well plates. After culturing for 24 h, cells were exposed continuously to the indicated drug concentrations without (○) or with 5 μM olaparib (●) for 24 h. Cells were immunostained for γH2AX and its accumulation in the nucleus was examined by immunofluorescence microscopy after counterstaining with 4’,6-diamidino-2-phenylindole. Each bar represents the mean±SE (n=3). Significant differences were determined by an ANOVA followed by Tukey's test (**p<0.01 vs. control without anticancer drug, ††p<0.01 vs. control with anticancer drug).
In conclusion, the PARP inhibitor, olaparib, synergistically potentiated the cytotoxicity of DNA damaging agents such as CDDP, doxorubicin, SN-38, and temozolomide by additively increasing DSBs in ESCC cell lines. Further research may be able to develop this finding into an effective therapeutic strategy against ESCC.

Effects of a DNA-dependent protein kinase inhibitor on cell growth inhibition by anticancer drugs plus olaparib in KYSE70 cells. Cells were seeded onto 96-well plates. After culturing for 24 h, cells were exposed continuously to the indicated drugs for one week without (○) or with 5 μM olaparib (●), or with olaparib plus NU7441 (▪). Cell viability was determined using the CellQuanti-Blue Cell Viability Assay Kit. Each point represents the mean±SE (n=4).
Acknowledgements
This study was supported, in part, by the JSPS KAKENHI grant number 16K18964.
Footnotes
Authors' Contributions
Study concept and design: T. Minegaki, K. Miyamoto. Acquisition of data: K. Miyamoto, M. Tanahashi, A. Yamamoto, Y. Moriyama, A. Wada, A. Matsumoto, K. Ota, M. Tanaka, U. Masuda. Analysis and interpretation of data: all Authors. Drafting of the manuscript: K. Miyamoto, T. Minegaki. Study supervision: M. Tsujimoto, K. Nishiguchi.
Conflicts of Interest
The Authors declare no conflicts of interest associated with this manuscript.
- Received January 22, 2019.
- Revision received February 22, 2019.
- Accepted February 26, 2019.
- Copyright© 2019, International Institute of Anticancer Research (Dr. George J. Delinasios), All rights reserved
References
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- DNA repair biomarkers to guide usage of combined PARP inhibitors and chemotherapy: a meta-analysis and systematic review
- Combining novel agents with radiotherapy for gynecologic malignancies: beyond the era of cisplatin
- Olaparib Potentiates Anticancer Drug Cytotoxicity via 53BP1 in Oesophageal Squamous Cell Carcinoma Cells