


Abstract
Background: The 90-kDa heat-shock protein (HSP90) is a chaperone protein expressed at high levels in cancer cells and is involved in the folding or stabilization of several client proteins, including epidermal growth factor receptor (EGFR). Ganetespib is a second-generation HSP90 inhibitor with a potent antitumor effect against various cancer types. Materials and Methods: This study examined the antitumor effect of ganetespib in EGFR-mutant non-small cell lung cancer (NSCLC) cells and experimentally established EGFR-tyrosine kinase inhibitor (TKI)-resistant cells harboring various resistance mechanisms, including EGFR T790M mutation, met proto-oncogene amplification, and epithelial–mesenchymal transition. Results: Ganetespib showed a potent antitumor effect at low concentrations, suppressing EGFR-related downstream pathway molecules and inducing cleavage of poly ADP-ribose polymerase in all examined EGFR-TKI-resistant cell lines in vitro. Ganetespib also inhibited in vivo tumor growth in resistant cells harboring EGFR T790M. Conclusion: Ganetespib might be a promising therapeutic option for the treatment of patients with EGFR-TKI-resistant NSCLC.
Lung cancer is the leading cause of cancer death worldwide (1). The frequency of epidermal growth factor receptor (EGFR) mutations among East Asian patients with non-small cell lung cancer (NSCLC) is approximately 30% (2), and recently developed molecular-targeted drugs, such as the EGFR-tyrosine kinase inhibitors (EGFR-TKIs) gefitinib and erlotinib, have improved the prognosis of patients with NSCLC who have EGFR mutations (3-5). However, almost all patients who initially respond to EGFR-TKIs eventually develop resistance within approximately 1 year of treatment. The most frequent resistance mechanism is EGFR T790M secondary mutation, followed by human epidermal growth factor receptor 2 (HER2) amplification, met proto-oncogene (MET) amplification, small-cell lung cancer transformation, epithelial–mesenchymal transition (EMT), and so on (6, 7). Thus, there is an urgent need for novel therapeutic strategies to overcome these resistance mechanisms.
The 90-kDa heat-shock protein (HSP90) is an ATP-dependent molecular chaperone that is highly expressed in cancer cells (8). The roles of HSP90 include ensuring appropriate folding or stabilization of client proteins such as EGFR, HER2, and anaplastic lymphoma kinase (ALK), which are oncogenic proteins that are essential for cancer cell growth, differentiation, and survival (9-11). The overexpression of HSP90 has been reported to be associated with poor survival in several cancer types (12-14). The inhibition of HSP90 results in rapid degradation of client proteins via the ubiquitin-proteasome system, simultaneously suppressing multiple oncogenic signaling pathways (9, 15). Thus, HSP90 is regarded as a new therapeutic target for cancer therapy (16). A number of HSP90 inhibitors have been developed, and preclinical studies have revealed their efficacy in a variety of cancer types, both in vitro and in vivo (17-20).
Characteristics and drug concentration required for 50% inhibition (IC50) of gefitinib and ganetespib in parental and experimentally established acquired epidermal growth factor receptor (EGFR)-tyrosine kinase inhibitor-resistant non-small cell lung cancer.
Ganetespib is a second-generation HSP90 inhibitor with improved pharmacological properties and safety profiles, such as reduced liver toxicity, compared to those of first-generation drugs (21). In some preclinical studies, ganetespib was also found to exhibit a potency superior to that of first-generation drugs (10, 11, 22). In clinical trials, when used as monotherapy or in combination with conventional chemotherapeutic agents or molecular targeting agents, ganetespib has been evaluated for a wide range of cancer types (21, 23-30). To our knowledge, however, only a few studies have investigated the effect of ganetespib against EGFR-TKI-resistant EGFR-mutant NSCLC cells (10, 11). Moreover, the effect of ganetespib in acquired EGFR-TKI resistance is unknown. In our previous study, we established several types of EGFR-TKI-resistant NSCLC cells with various resistance mechanisms, including EGFR T790M mutation, MET amplification and EMT (31, 32). The purpose of this study was to investigate whether ganetespib had an antitumor effect on EGFR-TKI-resistant NSCLC cells.
Materials and Methods
Cell culture and reagents. Four EGFR-mutant NSCLC cell lines in this study: HCCC827 (exon 19del E746-A750), PC-9 (exon 19del E746-A750), HCC4006 (exon 19del L747-E749), and HCC4011 (L858R). All the lines, except for HCC4011, were purchased from the American Type Culture Collection (Manassas, VA, USA). HCC4011 was kindly provided by Professor Adi F. Gazdar (University of Texas Southwestern Medical Center, Dallas, TX, USA). Five experimentally established EGFR-TKI-resistant cell lines, HCC827-GRS, resistant to gefitinib by stepwise escalation methods; HCC827-GRH1, resistant to gefitinib by high-concentration exposure methods; and PC-9-GRS, HCC4006-GRS, and HCC4011-GRS, which had acquired resistance to gefitinib by stepwise escalation, as reported previously (31). The resistance mechanisms of each drug-resistant cell are shown in Table I. All the cell lines were authenticated by short tandem repeat (STR) DNA analysis (Promega, Madison, WI, USA). All these cells were cultured in Roswell Park Memorial Institute-1640 medium supplemented with 10% fetal bovine serum at 37°C in a humidified incubator under 5% CO2 gas. Gefitinib was purchased from InvivoGen (San Diego, CA, USA), and ganetespib was purchased from ChemScene (Monmouth Junction, NJ, USA).
DNA extraction. Genomic DNA was extracted from the cell lines with a DNeasy Blood & Tissue Kit (Qiagen, Hilden, Germany).
Analysis of copy number by real-time polymerase chain reaction. Copy number gains of MET genes were determined by quantitative real-time polymerase chain reaction (qPCR) assay using TaqMan Genotyping Master Mix (Applied Biosystems, Foster City, CA, USA). The relative copy number of each sample was determined by comparing the ratio of the MET gene to that of glyceraldehyde 3-phosphate dehydrogenase in each sample with that of the MET gene in human genomic DNA (Promega, Madison, WI, USA).A copy number greater than 4 was considered as MET-amplified according to previous study (31, 33).
Direct sequencing. The mutational status of the EGFR gene was analyzed by direct sequencing. PCR conditions were as previously reported (31).
Cell growth-inhibition assay. The sensitivity to each drug was determined by a modified MTS assay (31). Cells (2×103 cells per well) were seeded in 96-well plates. After exposure to ganetespib for 72 h (four-fold serial dilutions with maximum concentration at 10 μM), 20 μl of CellTiter 96 AQueous One Solution Reagent (Promega) was added to each well. The antiproliferative activity of each drug are reported as the drug concentration required for 50% inhibition (IC50).
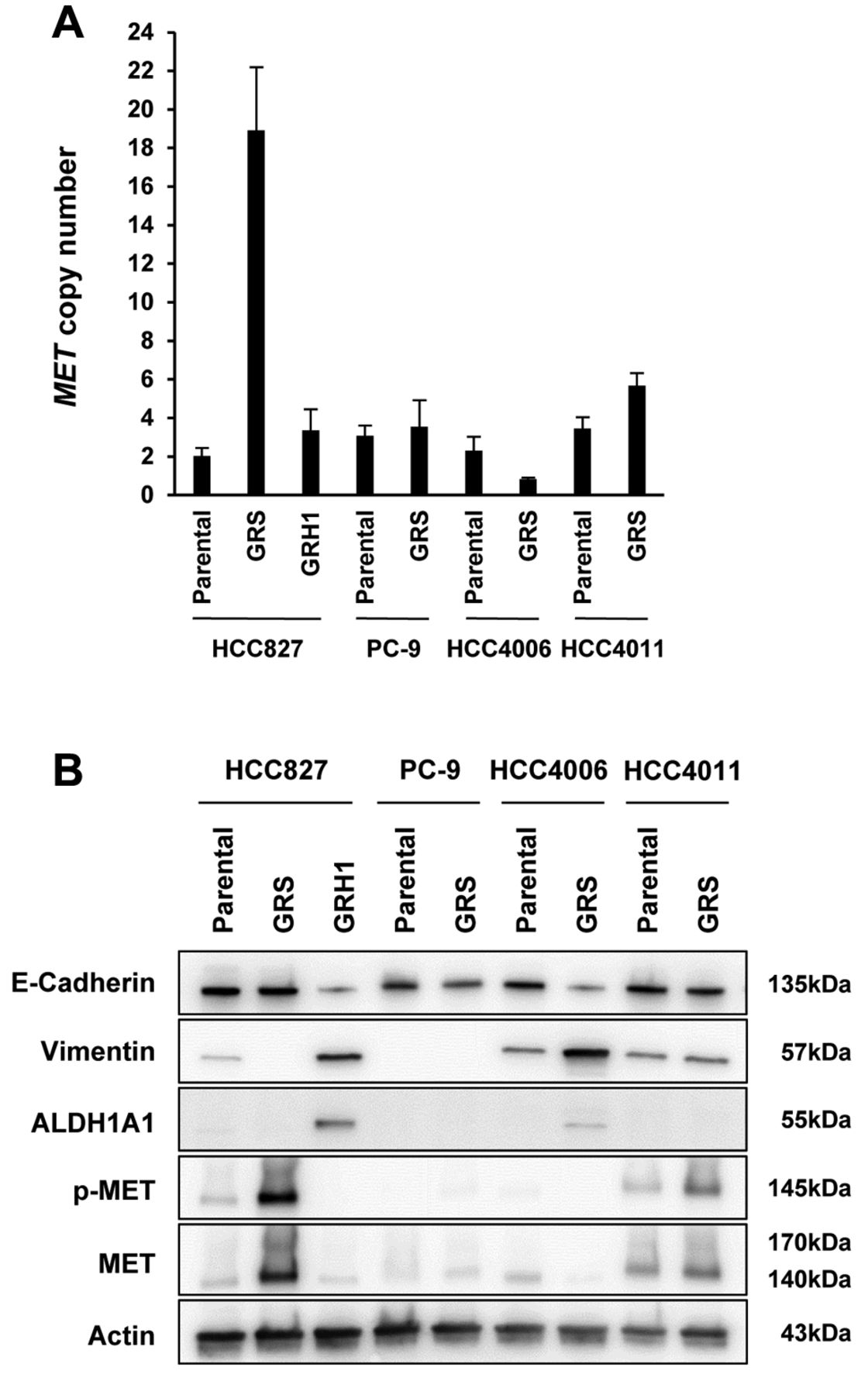
Mechanisms of acquired resistance in experimentally-established epidermal growth factor receptor (EGFR)-tyrosine kinase inhibitor-resistant non-small cell lung cancer cells. A: Met proto-oncogene (MET) copy number was increased in HCC827-GRS and HCC4011-GRS by quantitative polymerase chain reaction. B: In western blot, epithelial–mesenchymal transition features (down-regulation of epithelial marker E-cadherin and up-regulation of mesenchymal marker vimentin) and up-regulation of aldehyde dehydrogenase 1A1 (ALDH1A1) were observed in HCC827-GRH1 and HCC4006-GRS cells. In addition, total and phospho-MET were overexpressed in HCC827-GRS and HCC4011-GRS. GRS: Exposed to gefitinib by a stepwise escalation; GRH: exposed to gefitinib by high-concentration exposure.
Western blot analysis. All the cell lines were cultured in 10-cm dishes for 24 h, then treated with dimethyl sulfoxide (DMSO) (Wako, Osaka, Japan) as a control, and with 10, 20, and 100 nM ganetespib for 24 h. The total cell lysates were then extracted with lysis buffer, a mixture of radioimmunoprecipitation assay buffer (Sigma-Aldrich, St. Louis, MO, USA), phosphatase inhibitor cocktails 2 and 3 (Sigma-Aldrich), and complete Mini Protease Inhibitor Cocktail (Roche, Switzerland). The primary antibodies used were against EGFR, phospho-EGFR (Tyr1068), MET, phospho-MET (Tyr1234/1235), protein kinase B (AKT), phospho-AKT (Ser473), mitogen-activated protein kinase (MAPK), phospho-MAPK, E-cadherin, vimentin, aldehyde dehydrogenase 1A1 (ALDH1A1), poly ADP-ribose polymerase (PARP) (Cell Signaling Technology, Danvers, MA, USA), and β-actin (used as an equal loading control) (Merck Millipore, Burlington, MA, USA). The secondary antibodies were as follows: goat anti-mouse or anti-rabbit immunoglobulin G (IgG)-conjugated horseradish peroxidase (Cell Signaling). To detect specific signals, the membrane was examined using the ECL Prime Western Blotting Detection System (GE Healthcare, Amersham, UK) and LAS-3000 (Fujifilm, Tokyo, Japan). The relative band intensities were quantified using ImageJ software (National Institute of Health, Bethesda, MD, USA).
Xenograft model. This study was approved by the Institutional Animal Care and Use Committee (permission number: OKU-2018764). All the experimental animal procedures were performed in accordance with the Guidelines for the Care and Use of Laboratory Animals and all efforts were made to minimize animal suffering. We subcutaneously injected PC-9-GRS (2.5×106 cells) into the left flank of female BALB/c nude mice (6 weeks old; n=6 per group) purchased from Charles River Laboratories (Yokohama, Japan). Tumor volumes were calculated according to the formula V=1/2ab2 (a: the long diameter, b: the short diameter of the tumor). When the mean tumor volume reached 100-150 mm3, the mice were treated with 45 mg/kg of ganetespib by intraperitoneal injection in the diluent 10/18 DRD [10% DMSO, 18% Cremophor RH40 (Sigma-Aldrich), 3.6% dextrose (Sigma-Aldrich), and 68.4% water] or with vehicle control (10, 19). Tumor volumes were measured three times a week with an electric caliper, as well as body weight. After 4 weeks of treatment, the mice were euthanized and the tumors were removed, measured and photographed.
Statistical analysis. The statistical analysis was performed using the GraphPad Prism (GraphPad Software, San Diego, CA, USA). Data from two groups were compared using Student's t-test. Differences with values of p<0.05 were considered to be statistically significant. All tests were two-sided. The results are presented as the mean±standard error (SE).
Results
Mechanisms of acquired resistance in experimentally established EGFR-TKI-resistant NSCLC cells. All examined EGFR-TKI (gefitinib)-resistant cells had been established previously in our group, with the resistance mechanisms consisting of EGFR T790M, MET amplification and EMT (31) (Table I). At first, we confirmed the characteristics of EGFR-TKI-resistant cell lines. EGFR T790M secondary mutations were detected in PC-9-GRS by direct sequencing of genomic DNA (data not shown). The MET copy number was increased in HCC827-GRS and HCC4011-GRS by qPCR (Figure 1A), and total and phospho-MET were also overexpressed in these cell lines compared with parental cell lines by western blot analysis (Figure 1B). EMT features were observed in HCC827-GRH1 and HCC4006-GRS with down-regulation of epithelial marker E-cadherin and up-regulation of mesenchymal marker vimentin (Figure 1B). In addition, up-regulation of cancer stem cell-related marker ALDH1A1 was also observed in HCC827-GRH1 (Figure 1B). As a result, we confirmed that the resistant cells retained their characteristics as the previous report (31).

Ganetespib exhibits a potent antitumor effect on parental non-small cell lung cancer cells and cells with acquired resistance (GRS: exposed to gefitinib by stepwise escalation; GRH: exposed to gefitinib by high-concentration exposure). Cells were treated with the indicated concentrations of gefitinib or ganetespib for 72 h. Cell viability was determined by the MTS assay.
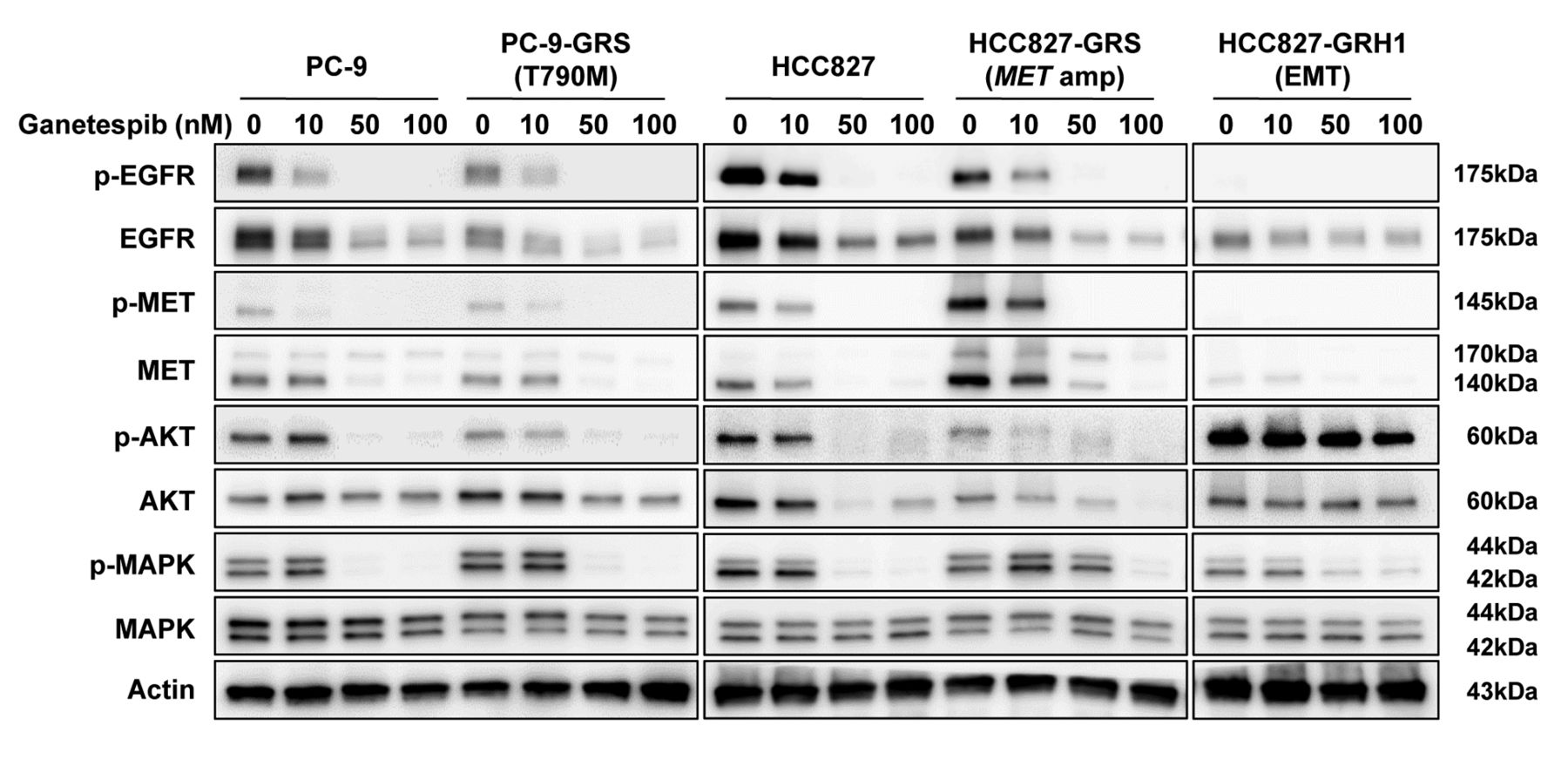
Ganetespib suppresses epidermal growth factor receptor (EGFR) and its related downstream pathway molecules in non-small cell lung cancer cells with acquired resistance to EGFR-tyrosine kinase inhibitor. Cells were treated with the indicated concentration of ganetespib for 24 h, and lysates were analyzed by western blot. AKT: protein kinase B; EMT: epithelial–mesenchymal transition; MET: met proto-oncogene; amp: amplification; MAPK: mitogen-activated protein kinase.
Ganetespib exhibited a potent antitumor effect on EGFR-TKI-resistant NSCLC cells regardless of the resistance mechanisms. The IC50 concentrations for gefitinib and ganetespib against each parental NSCLC cell and each acquired EGFR-TKI-resistant cell are shown in Table I and Figure 2. The IC50 values for ganetespib in parental NSCLC cells ranged from 5.9 to 22 nM, and those in acquired EGFR-TKI-resistant cells ranged from 5.4 to 31 nM. Thus, ganetespib demonstrated a potent antitumor effect on acquired EGFR-TKI-resistant NSCLC cells regardless of their resistance mechanisms (Figure 2E–H).
Ganetespib suppressed EGFR-related downstream pathway molecules in NCSLC cells with acquired EGFR-TKI resistance. Western blot analyses were performed to investigate alterations in downstream pathway molecules, such as both the total and the phosphorylated EGFR, MET, AKT, and MAPK after ganetespib treatment. Cells were harvested after 24 h of treatment with 0, 10, 50, and 100 nM of ganetespib. Ganetespib suppressed expression of EGFR and its related downstream pathway molecules in parental and acquired EGFR-TKI-resistant NSCLC cells harboring EGFR T790M, MET amplification, and EMT (Figure 3).
Ganetespib induced cleavage of PARP in NCSLC cells with acquired EGFR-TKI resistance. We analyzed the expression of the apoptosis marker cleaved PARP after ganetespib treatment. Cells were harvested after 72-h treatment with 0, 10, and 50 nM ganetespib. Ganetespib induced cleavage of PARP in parental NSCLC cells and cells with acquired EGFR-TKI resistance (Figure 4).
Ganetespib inhibited tumor growth of NCSLC cells with acquired EGFR-TKI resistance in a mouse xenograft model. We evaluated the antitumor effect of ganetespib in a PC-9-GRS mouse xenograft model. We selected PC-9-GRS because its resistance mechanism, EGFR T790M, is the most frequent mechanism of EGFR-TKI resistance. The dose of ganetespib was determined according to previous reports (10, 34, 35). As shown in Figure 5A, ganetespib treatment significantly inhibited the tumor growth of mouse xenografts bearing PC-9-GRS compared with the vehicle control at 4 weeks after the beginning of the treatment (p<0.01). Moreover, ganetespib treatment did not lead to significant body weight loss (Figure 5B).
Discussion
Although HSP90 inhibitors bind to only one target, HSP90, they simultaneously block various client proteins involved in multiple oncogenic signaling pathways (11). Therefore, they are regarded as promising therapeutics for cancer treatment. Ganetespib is a second-generation HSP90 inhibitor with improved pharmacological properties and safety profiles compared to those of first-generation drugs (21). Rouhi et al. reported Kirsten rat sarcoma viral oncogene homolog (KRAS)-mutant colorectal cancer cells that acquire resistance by overexpression of the drug efflux pump ATP-binding cassette subfamily B member 1 (ABCB1) were sensitive to ganetespib, whereas they had been insensitive to the first-generation HSP90 inhibitor 17-allylamino-17-demethoxy-geldanamycin (17-AAG). One possible explanation is that 17-AAG is exported by ABCB1; however, findings have revealed that ganetespib might not be a substrate of ABCB1, thus, it is not exported (36).

Ganetespib induces cleavage of poly ADP-ribose polymerase (PARP) in non-small cell lung cancer cells with acquired resistance to epidermal growth factor receptor (EGFR)-tyrosine kinase inhibitor. Cells were treated with the indicated concentrations of ganetespib for 72 h, and lysates were analyzed by western blot. The relative band intensity of cleaved PARP (quantified by densiometric analysis using ImageJ software) was up-regulated by ganetespib treatment. EMT: Epithelial–mesenchymal transition; MET: met proto-oncogene; amp: amplification.

Ganetespib inhibits growth of xenografts of non-small cell lung cancer cells harboring the epidermal growth factor receptor (EGFR) T790M mutation (PC-9-GRS) with acquired resistance to EGFR-tyrosine kinase inhibitor A: Tumor volume was measured three times a week. **Significantly different at p<0.01. B: Body weight was measured three times a week. The percentage of body weight compared to that of day 0 is shown. The body weight was not significantly affected by ganetespib (p=0.41). Data are shown as mean±SE (n=6). GRS: Exposed to gefitinib by stepwise escalation.
In our study, we showed the potent antitumor effect of ganetespib on three types of NSCLC cells with acquired EGFR-TKI resistance, EGFR T790M, MET amplification, and EMT. EGFR suppression and its related downstream pathway molecules and induction of cleavage of PARP were believed to contribute to its efficacy. We focused on EGFR T790M, which is the main cause of acquired EGFR-TKI resistance, and demonstrated strong tumor growth inhibition by ganetespib treatment in vivo.
As described earlier, ganetespib has a great efficacy on cancer cells, including EGFR-TKI-resistant NSCLC cells; however, Chatterjee et al. established acquired ganetespib-resistant cells in KRAS-mutant NSCLC cells (37). They identified an extracellular signal-regulated kinase downstream target, p90 ribosomal S6 kinase (RSK), as a central mediator of resistance and suggested that the combination of ganetespib with an RSK inhibitor can overcome acquired ganetespib resistance. Investigating the mechanism of acquired resistance to ganetespib of EGFR-mutant NSCLC cells would be necessary in future studies.
In terms of clinical studies, the phase III GALAXY-2 trial was the most advanced study on ganetespib to date. This trial compared ganetespib and docetaxel to docetaxel alone in patients with advanced NSCLC who had already received one prior systemic therapy for advanced therapy. Although, contrary to their expectation, ganetespib did not improve overall or progression-free survival, this study had excluded patients with EGFR-mutant NSCLC. Based on the results of our data and this clinical trial, appropriate patient selection (e.g. patients with acquired EGFR-TKI resistance) might be important to demonstrate the effect of ganetespib in some subsets of patients with NSCLC.
Our study had several limitations. EGFRF-TKI-resistant NSCLC cell lines harboring MET amplification or EMT features did not engraft well enough in the mouse xenograft model, and we were therefore unable to demonstrate the effect of ganetespib against these cell lines in vivo.
In conclusion, we showed that ganetespib exhibits potent in vivo and in vitro antitumor effects against NSCLC cells with various mechanisms of acquired EGFR-TKI resistance. These findings suggest that ganetespib might be a promising therapeutic option for the treatment of patients with EGFR-TKI-resistant NSCLC.
Acknowledgements
The Authors thank Ms. Fumiko Isobe (Department of General Thoracic Surgery and Breast and Endocrinological Surgery, Okayama University Graduate School of Medicine, Dentistry and Pharmaceutical Sciences, Okayama, Japan) for her technical support. This work was supported by a Management Expenses Grant and a Grant-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (JSPS KAKENHI grant numbers 17K16608 to K. Shien; 16H05431 to S. Toyooka).
Footnotes
Authors' Contributions
Eisuke Kurihara performed majority of the experiments and wrote the article. Kazuhiko Shien designed the research and helped to write the article. Hidejiro Torigoe, Takahiro Yoshioka, Kei Namba, and Hiroki Sato contributed to in vitro experiments. Tatsuaki Takeda, Yuta Takahashi, and Yuske Ogoshi participated in the animal experiments. Ken Suzawa and Shuta Tomida performed the statistical analysis. Hiromasa Yamamoto, Junichi Soh, Mikio Okazaki, and Tadahiko Shien contributed to analysis and interpretation of data, and reviewed the article. Shinichi Toyooka supervised this study.
This article is freely accessible online.
Conflicts of Interest
The Authors have no conflicts of interest directly relevant to the content of this article.
- Received February 8, 2019.
- Revision received March 3, 2019.
- Accepted March 7, 2019.
- Copyright© 2019, International Institute of Anticancer Research (Dr. George J. Delinasios), All rights reserved
References
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.