


Abstract
Background/Aim: PLX4032 is commonly used in the treatment of advanced melanoma patients with BRAF-V600E mutation. The aim of this study was to elucidate the mechanisms by which up-regulation of PIN1 confers PLX4032 resistance in melanoma. Materials and Methods: The expression of PIN1 as well as the cytotoxic effects of combinatorial treatment of PLX4032 and all-trans retinoic acid (ATRA) were investigated by immunoblotting, MTT assay, TUNEL assay, and soft agar assay. Results: PIN1 expression is up-regulated in A375R cells, a PLX4032-resistant subline of melanoma cells generated from an A375 cell line, compared to parental A375 cells. Indeed, PIN1 positively regulated the expression of EGFR in A375R cells and led to activation of the RAF/MEK/ERK pathway. Importantly, PLX4032, when used in combination with ATRA, an inhibitor of PIN1, reduced EGFR expression, and consequently reduced cell viability and anchorage-independent growth of A375R cells compared to PLX4032 alone. Furthermore, co-treatment with ATRA and PLX4032 increased cleaved PARP and DNA fragmentation in A375R cells. Conclusion: PIN1 plays an important role in the development of PLX4032 resistance through up-regulation of EGFR expression.
Melanoma is a highly malignant form of skin cancer that develops from pigment-containing cells termed melanocytes (1, 2). A recent report indicated that melanoma comprises approximately 232,100 (1.7%) cases of all newly-diagnosed primary malignant cancer, and causes about 55,500 deaths each year worldwide (2). Compared to other types of skin cancers such as basal cell carcinoma and squamous cell carcinoma, melanoma is more likely to metastasize, and tends to become life-threatening rapidly after metastasis (2). Epidemiological surveys have revealed that the incidence of melanoma in most developed countries has risen faster than any other cancer type since the mid-1950s (3, 4). Melanoma is classified into 4 subtypes on the basis of activating gene mutations, which include: BRAF mutant melanoma, Ras-mutant melanoma, NF-1 mutant melanoma and triple wild-type melanoma. BRAF mutations are the most predominant activating mutations in melanoma, and occur in approximately 50% of cases (2, 5, 6). Among the BRAF mutations present in melanoma, about 90% involve a single nucleotide mutation at codon 600, resulting in substitution of glutamic acid for valine (BRAF V600E, nucleotide 1799 T>A; codon GTG>GAG) (7). BRAF V600E mutant kinase is constitutively active as well as insensitive to negative feedback mechanisms, which leads to the sustained activation of the RAS/RAF/MEK signaling pathway, independent of upstream regulators (8). Up-regulation of RAS/RAF/MEK signaling is implicated in different mechanisms of melanoma progression such as unrestricted cell proliferation, evasion of apoptosis, and angiogenesis (8). Advanced melanoma patients with BRAF V600E mutation are treated with PLX40320, which has been shown to be very effective compared to high dose IL-2 and dacarbazine, the only therapies approved by the FDA for the treatment of metastatic melanoma prior to the approval of PLX4032 (9, 10). However, the clinical efficacy of PLX4032 is often temporarily due to the rapid development of acquired drug resistance.
Several mechanisms have been reported to be involved in the development of PLX40320 resistance in melanoma, including activating mutations in N-Ras and MEK, amplification and alternative splicing of the BRAF gene, and upregulation of EGFR, C-RAF and MAP3K8 (11-13). Among several mechanisms responsible for resistance, it has been suggested that most tumors that initially respond to BRAF inhibitors eventually develop acquired resistance through activation of alternative pathways leading to reactivation of cell proliferation (14, 15). Indeed, the exposure of colorectal cancer cells to BRAF inhibitors results in a feedback activation of EGFR and lack of sensitivity to PLX4032 (11). Up-regulation of EGFR is most commonly observed in PLX40320-resistant melanoma, which leads to reactivation of RAF/MEK/ERK and/or PI3K/AKT signaling to promote resistance (12). Thus, inhibition of EGFR signaling by monoclonal antibody (i.e., cetuximab) or tyrosine kinase inhibitors (TKI) (i.e., erlotinib, gefitinib and lapatinib) is synergistic with BRAF inhibition in colon carcinoma cells (16). Indeed, the pan-EGFR TKI, lapatinib, prevents ERK reactivation and sensitizes BRAF-mutant thyroid cancer cells to RAF or MAPK kinase inhibitors (17). These findings elucidate the important role of EGFR in mediating PLX4032 resistance, suggesting that inhibition of EGFR expression might be a potential therapeutic approach to overcome resistance. However, mechanisms underlying the expression of EGFR in PLX4032-resistant melanoma cells are poorly understood.
PIN1 is the only known peptidyl-prolyl isomerase that binds and isomerizes specific phosphorylated serine or threonine residues preceded by proline (18-20). PIN1-induced conformational changes have profound effects on the function of many substrate proteins by modulating their structure, de-phosphorylation, protein–protein interactions, subcellular localization, and protein stability (18). PIN1 has been shown to be involved in the regulation of many cellular events, such as cell-cycle progression, transcriptional modulation, and cell proliferation (18). Furthermore, PIN1 is highly overexpressed in many human cancers, and high PIN1 levels correlate with poor prognosis in breast cancer (19, 20). Previous reports indicated that PIN1, through its interaction with MEK1, enhances HER2 expression, a closely related receptor tyrosine kinase member of the EGFR family of receptors (21). However, it is not known whether PIN1 regulates EGFR expression, resulting in acquisition of PLX4032 resistance in melanoma. A recent study showed that retinoid derivative ATRA, which is already approved for the chemotherapy of acute myeloid leukemia (AML), is a strong inhibitor of PIN1 (22), suggesting a potential use of ATRA in the treatment of PLX4032-reisistant melanoma. Herein, we showed that EGFR expression is positively regulated by PIN1 and suggest that inhibition of PIN1 using ATRA may enhance PLX4032 sensitivity in resistant melanoma. Our findings illustrate a novel strategy to recover from PLX4032 resistance by intervening in EGFR expression, and also expand understanding of the role of PIN1 in PLX4032 resistance in melanoma.
Materials and Methods
Cell culture and establishment of resistant cell line. A375 cells, purchased from the American Type Culture Collection (ATCC), were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) at 37°C in humidified air containing 5% CO2. To establish the A375R cells, stepwise drug selection was continued until the A375 cell population could sustain viability and proliferation when challenged with 10 μM of PLX4032. The established A375R cells were maintained in DMEM with 10% FBS and 10 μM PLX4032.
Antibodies and reagents. Antibodies against p-MEK1/2 (1:1,000), p-ERK1/2 (1:1,000), cyclin D1 (1:1,000), p-RAF1(1:1,000), and cleaved caspase 3 (1:1,000) were purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA); antibodies against EGFR (1:1,000) and cleaved PARP (1:5,000) were from Santa Cruz Biotechnology (Dallas, TX, USA); β-actin (1:10,000) was from Sigma-Aldrich (St. Louis, MO, USA); anti-Xpress (1:5,000) was from Invitrogen (Waltham, MA, USA). Reagents against ATRA was purchased from Sigma-Aldrich (St. Louis, MO, USA). PD98059 was purchased from Calbilchem-Novabiochem (San Diego, CA, USA). PLX 4032 was obtained from Advanced ChemBlock (San Diego, CA, USA). The jetPEI® cationic polymer transfection reagent was from Polyplus-transfection (Illkrich, France). Lipofectamine Plus reagent transfection reagent was from Invitrogen (Grand Island, NY, USA).
Mammalian expression vectors and small interfering RNA. The cDNA of full sequence of PIN1, a gift from Dr. Kun Ping Lu, was subcloned into the pcDNA4/Xpress vector (Invitrogen). The silencing of human PIN1 (accession number: NM_005221) was carried out by transfecting the ON-TARGETplus siRNA SMART pool-specific or nonspecific control pool double-stranded RNA oligonucleotides (Dharmacon, Chicago, IL, USA) using Lipofectamine 2000 (Invitrogen).
MTT assays. MTT assays were performed to check cell viability using EZ-Cytox Cell viability assay kit (Daeli Lab Service, Seoul, Republic of Korea). In brief, cells (1×104) were seeded in 96-well plates with 100 μl of cell suspension in each well, and incubated at 37°C in humidified air containing 5% CO2. After 24 h in culture, cells were incubated with different concentrations and combination of drugs for 48 h. The cells were then treated with 10 μl of tetrazolium salt and incubated for 4 h. The absorbance due to purple formazan formed by live cells was measured at 450 nm.
Protein immunoblotting. For immunoblotting, cells grown in monolayers were harvested, washed in PBS, and lysed in RIPA buffer containing 150 mM NaCl, 50 mM Tris-HCl (pH 7.4), 0.25% sodium deoxycholate, 1 mM EDTA, 1% NP40, 1 mM NaF, 0.2 mM PMSF, 0.1 mM sodium orthovanadate, and protease inhibitor cocktail (Roche Life Science, Indianapolis, IN, USA). Protein samples were resolved using SDS-PAGE and blotted onto PVDF membranes, and immunoblots were probed with the indicated antibodies. The immunoblots were visualized using a SuperSignal West Femto chemiluminesecene substrate (ThermoScientific, Waltham, MA, USA) and detected by a LAS4000-mini biomolecular imager (FUJIFILM, Tokyo, Japan).
Anchorage-independent cell transformation (soft agar assay). Briefly, cells (8×103) were exposed to the indicated drug in 1 ml of 0.3% Eagle's basal medium (BME) containing 10% FBS. The cultures were maintained at 37°C in a 5% CO2 incubator for 14 days. Resultant cell colonies were scored using an Axiovert 200M fluorescence microscope and Axio Vision software (Carl Zeiss, Thornwood, NY, USA).
TdT-mediated dUTP nick end-labeling (TUNEL) assay. The induction of apoptosis was assessed by TUNEL staining and detected with an in situ Cell Death detection kit (Roche Life Science), according to the manufacturer's instructions. Briefly, 2×105 cells were cultured for 24 h in six-well plates. The cells were then starved for 24 h, then treated with PLX4032 or ATRA for 24 h. Treated cells were washed with PBS and fixed with Cytofix/Cytoperm™ (BD Biosciences, San Diego, CA, USA) at 4°C for 20 min. Cells were stained with 50 μl TUNEL solution at 37°C for 1 h, then washed twice with PBS and fixed. DNA fragmentation was detected using an Axiovert 200 M fluorescence microscope and quantified using Axiovision software (Carl Zeiss).
Cell-cycle analysis. A375 and A375R cells were seeded and treated with PLX4032 or ATRA, alone or in combination, respectively. The cells were washed, fixed with 70% ethanol, and then 200 μl of Muse™ Cell cycle reagent (EMD Millipore Corp. Billerica, MA, USA) was added. Then, cells were incubated at 25°C for 30 min in the dark. Samples were measured with a Muse Cell Cycle Assay kit (Merck Millipore, Billerica, MA, USA).
Xenografts and treatment. Male BABL/C mice (6 weeks) were obtained from the Orient (Seongnam, Republic of Korea) and maintained cages in a light and temperature-controlled room. A375 or A375R cells were subcutaneously injected (2×106) on mice flanks. After day 5, mice were randomly divided in to different groups. Mice were intraperitoneally injected three times a week with Saline (n=8), PLX4032 (20 mg/kg) (n=8) or combination of PLX4032 (20 mg/kg) and ATRA (10 mg/kg) (n=8). Mice were euthanized at 25 days after the first injection and calculated tumor volume and weight in all group. The tumor volume was measured using the callipers (Mitutoyo, Kawasaki, Japan) and the volumes was calculated as follows: tumor volume=0.5× [(large diameter) ×(small diameter)2]. All animal care and experimental procedures complied with local guidelines and were approved by the Animal Experiments Committee of Chosun University.
Statistical analysis. Data from the MTT assays, soft agar assays, sub-G1 assays, and TUNEL assays were statistically analyzed using unpaired Student's t-tests, and p-values<0.05, 0.01, or 0.001 were considered significant.
Results
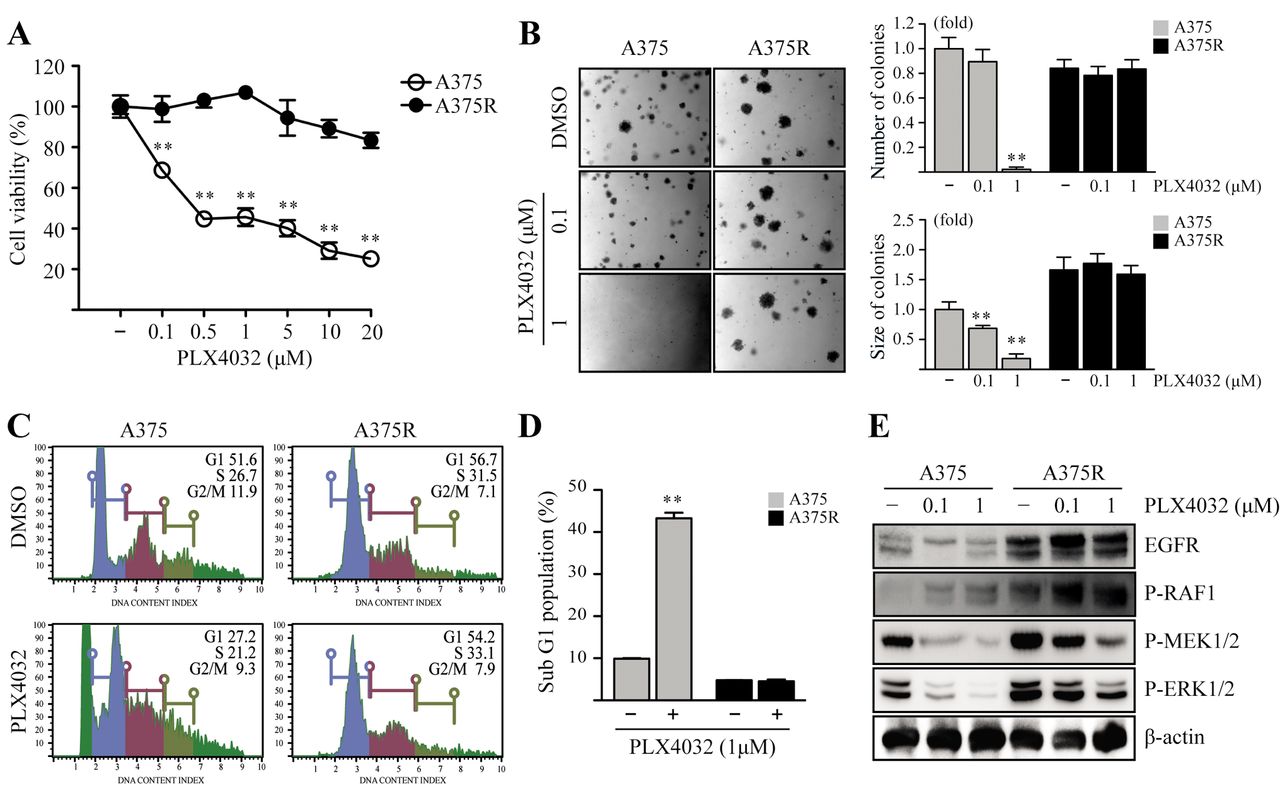
Increased EGFR levels are associated with resistance to PLX4032 in A375 cells. To study the mechanisms of resistance to PLX4032 in melanoma, we established a PLX4032-resistant melanoma cell line, designated as A375R, by continuous exposure of A375 cells to increasing concentrations (up to 10 μM) of PLX4032. Cell viability assays showed that the A375R cells were resistant to PLX4032 (IC50>10 μM) compared to parental A375 cells (IC50=0.8 μM) (Figure 1A). Consistent with these results, we found that A375R cells displayed considerable resistance to PLX4032 treatment, as evident by enhanced colony formation compared to A375 cells in soft agar assays (Figure 1B). We then studied cell-cycle distribution using a fluorescence-based cell cycle analyzer. As expected, sub-G1 population of A375 cells was increased from 9.9% to 43.3% with simultaneous decrease in G1 phase upon PLX4032 treatment, whereas A375R cells were able to abrogate the PLX4032-induced accumulation of sub-G1 population (Figure 1C-D). As previous reports indicate that reactivation of the MAPK signaling pathway is responsible for the development of PLX4032 resistance (23), we examined the protein levels of EGFR and activation of the RAF1/MEK1/2/ERK1/2 signaling pathway in A375 and A375R cells by immunoblotting. It was observed that treatment of parental A375 cells with PLX4032 caused a robust dose-dependent inhibition of MEK1/2 and ERK1/2 activities, and also decreased EGFR levels (Figure 1E). However, EGFR expression and levels of phosphorylated MEK1/2 and ERK1/2 remained relatively unchanged in A375R cells, even in the presence of vemurafenib (Figure 1E). Taken together, these results suggest that reactivation of the RAF1/MEK1/2/ERK1/2 pathway is responsible for enhanced survival and anchorage-independent growth of PLX4032 resistant A375R cells.
PIN1 mediates PLX4032 resistance through activation of RAF1-MEK1/2-ERK1/2 signaling in A375R cells. Given that PIN1 binds with MEK and promotes chemoresistance in breast cancer cells (24, 25), we hypothesized that PIN1 plays a role in the development of PLX4032 resistance in melanoma cells. To determine the possible association of PIN1 with PLX4032 resistance, we first compared the protein levels of PIN1 in A375 and A375R cells by immunoblotting. Results showed that PIN1 levels were elevated in A375R cells, and this positively correlated with the expression of EGFR and with the levels of phosphorylated RAF1, MEK1/2 and ERK1/2 (Figure 2A). To further examine the role of PIN1 in mediating resistance to PLX4032, we overexpressed PIN1 in A375 cells. This caused a significant increase in EGFR levels, with a concomitant increase in the phosphorylation of RAF1, MEK1/2, and ERK1/2 (Figure 2B). In contrast, PIN1 silencing in A375R cells decreased EGFR levels and attenuated the phosphorylation of RAF1, MEK1/2, and ERK1/2 (Figure 2C). Furthermore, PIN1 overexpression in A375 cells decreased the sensitivity to PLX4032, whereas PIN1 knockdown in A375R cells restored sensitivity to increasing doses of PLX4032, as shown by EGFR expression and activation of RAF1-MEK1/2-ERK1/2 signaling (Figure 2D and E). Taken together, these results suggest that PIN1 positively regulates the development of PLX4032 resistance in melanoma through the up-regulation of EGFR expression.
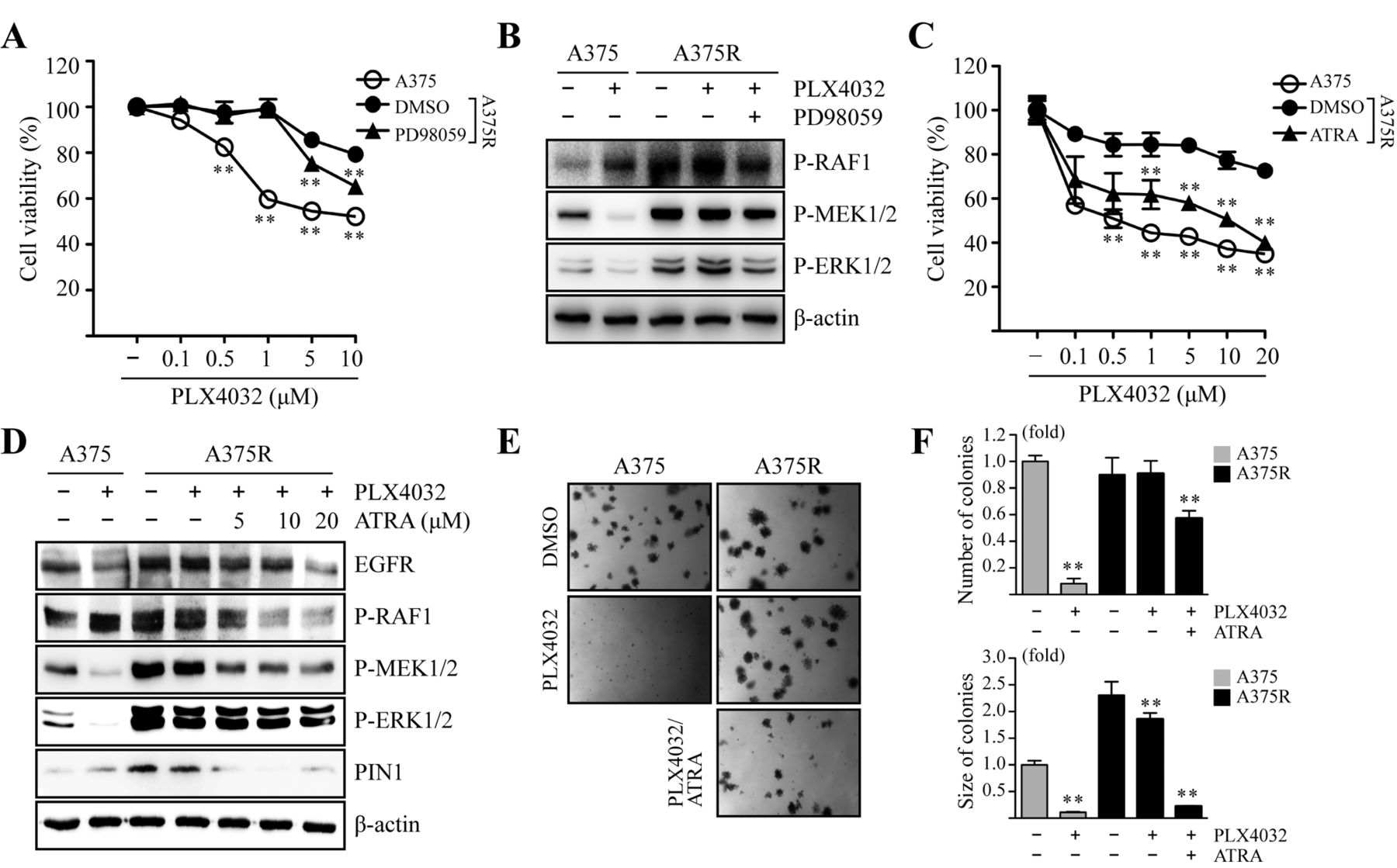
PIN1 inhibition by ATRA restores PLX4032 sensitivity via down-regulation of RAF1-MEK1/2-ERK1/2 signaling in A375R cells. The increased PIN1 and EGFR expression in PLX4302-resistant melanoma cells (Figure 2A) supports the hypothesis that such resistance may occur through activation of growth factor signaling via ERK1/2 signaling (26). Therefore, we examined whether treatment of PLX4032 with MEK inhibitor PD98059 in combination increased PLX4032 sensitivity in A375R cells. The co-treatment of PD98059 with PLX4032 did not significantly improve the PLX4032 sensitivity of A375R cells in terms of cell viability (Figure 3A) and RAF1-MEK1/2-ERK1/2 signaling (Figure 3B). Next, the role of PIN1 in conferring resistance to PLX4032 treatment lead us to speculate whether it is possible to recover such resistance by treatment with ATRA, a potent PIN1 inhibitor. Therefore, we performed cell viability assays to examine the growth inhibitory effects of ATRA on A375R cells. Notably, treatment with a combination of PLX4032 and ATRA in A375R cells was more effective than PLX4032 alone in reducing cell viability (Figure 3C). Next, we measured the levels of phosphorylated RAF1, MEK1/2 and ERK1/2 by immunoblotting after treatment with ATRA. Results showed that co-treatment of PLX4032 and ATRA in A375R cells reduced the levels of EGFR, as well as phosphorylated RAF1, MEK1/2 and ERK1/2 (Figure 3D), suggesting the down-regulation of MAPK signaling pathway by PIN1 inhibition. Concomitant with these results, treatment with PLX4032 and ATRA in combination also reduced the anchorage-independent colony-forming capacity of A375R cells (Figures 3E and F). Taken together, these results suggested that suppression of PIN1 may overcome the resistance of cells to PLX4032 for the treatment of melanoma through the downregulation of the RAF1-MEK1/2-ERK1/2 signaling pathway.

PLX4032-resistant cells display enhanced MAPK pathway activation. (A) A375 and A375R cells were seeded and cultured for 24 h at 37°C in a 5% CO2 atmosphere. Then, the cells were treated with various concentrations of PLX4032, as indicated. Cell viability was estimated using MTT assays. Points, mean of three experiments; bars, S.D.; **p<0.01. (B) A375 and A375R cells were exposed to the indicated concentrations of PLX4032 in soft agar matrix and incubated at 37°C in a 5% CO2 atmosphere for 14 days. Colonies from three independent experiments are shown. The average number of colonies was calculated, and colony sizes were measured under a microscope. Columns, mean of three experiments; bars, S.D.; **p<0.01. (C) A375 and A375R cells were treated with PLX4032. After 24 h, cells were harvested and used for cell-cycle analysis, as described in materials and methods. (D) A375 and A375R cells were seeded and treated with PLX4032, as indicated, for 24 h. Cells were harvested and proteins in whole-cell lysates were separated by SDS-PAGE and immunoblotted.
PIN1 inhibition by ATRA enhances PLX4032-induced apoptotic signaling in A375R cells. As PIN1 inhibition inhibited the RAF1-MEK1/2-ERK1/2 signaling pathway, we next assessed the effects of ATRA and PLX4032 in combination, on apoptosis of PLX4032-resistant A375R cells. To measure apoptosis, we detected cleaved caspase 3 and PARP levels by immunoblotting. Co-treatment with PLX4032 and ATRA induced greater cleavage of caspases 3 and PARP in A375R cells, compared with the PLX4032-only treated group (Figure 4A). To determine whether the combination of PLX4032 and ATRA also influences the progression of cell cycle, we performed cell-cycle analysis. As a result, the co-treatment of PLX4032 and ATRA led to a strong accumulation of the sub-G1 population in A375R cells with concomitant decrease in G1, S, and G2/M phase, compared to vehicle or PLX4032-only treated A375R cells (Figure 4B-C). Consistent with these results, terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick end labeling (TUNEL) staining revealed much more extensive nuclear fragmentation of cells in the PLX4032 and ATRA co-treated group of A375R cells, compared with only PLX4032-treated groups (Figure 4D). These results suggested that PIN1 inhibition restores the pro-apoptotic and anti-proliferative effects of PLX4032 in PLX4032-resistant melanoma.
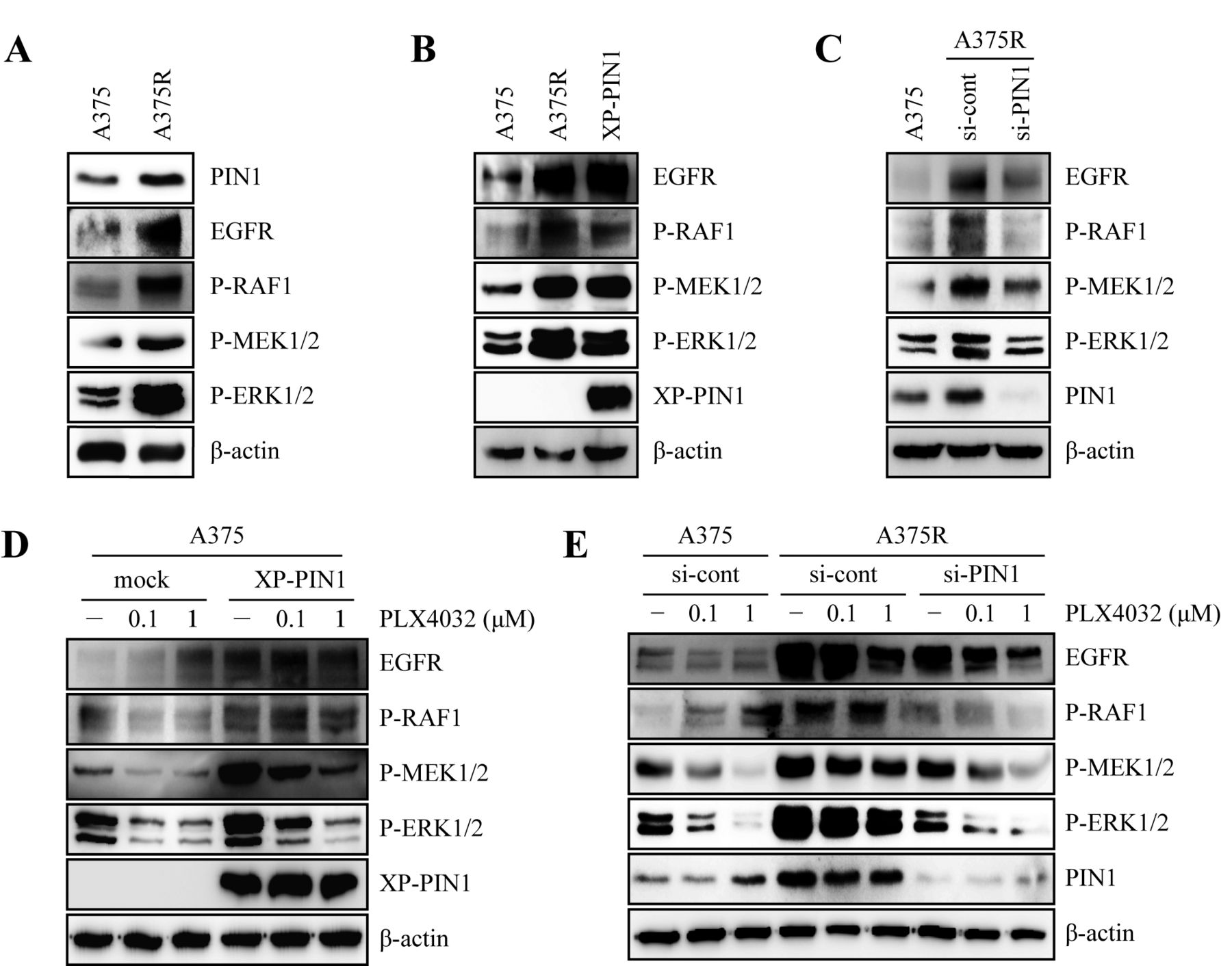
PIN1 overexpression is associated with PLX4032 resistance in human melanoma. (A) A375 and A375R cells were seeded and cultured for 48 h. Then, the cells were harvested, lysed and immunoblotted using relevant antibodies. (B) A375 cells were transfected with XP-PIN1 and incubated for 48 h. Then, the cells were harvested, lysed and immunoblotted using appropriate antibodies. (C) A375R cells were transfected with siRNA-control or siRNA-PIN1. After 48 h, the cells were harvested, lysed and immunoblotted using relevant antibodies. (D) A375 cells were seeded and transfected with mock or XP-PIN1. At 24 h after transfection, the cells were treated with the indicated concentrations of PLX4032 for 24 h, harvested, lysed and then immunoblotted using appropriate antibodies. (E) A375 and A375R cells were seeded and transfected with siRNA-control or siRNA-PIN1.
Co-treatment with PLX4032 and ATRA enhances PLX4032 sensitivity in vivo. To demonstrate the roles of PIN1 and PLX4032 resistance in melanoma cells in vivo, the anti-tumor effects of PLX4032 and ATRA in combination on A375R cell growth were examined in a murine melanoma-xenograft model. The growth of engrafted tumors was significantly increased in mice implanted with A375R cells compared with control cells (A375) (Figure 5A-C). Furthermore, tumor volumes and weights in A375R mice treated with PLX4032 and ATRA in combination were significantly lower than those in vehicle-treated A375R mice, whereas tumors were not decreased in the PLX4032 single-treated A375R mice. (Figure 5A-C). These data strongly support the concept that the RAF1-MEK1/2-ERK1/2 axis induced by PIN1 increases PLX4032 resistance in melanoma.

Treatment with PLX4032 and ATRA in combination increases PLX4032 sensitivity in A375R cells. (A) A375 and A375R cells were treated with the indicated concentrations of PLX4032 or PD98059 (10 μM), alone or in combination for 24 h, after which cell viability was measured via MTT assays. Points, mean of three experiments; bars, S.D.; **p<0.01. (B) A375 and A375R cells were treated with PLX4032 (1 μM) or PD98059 (10 μM), alone or in combination, for 24 h, then the cells were harvested and whole cell lysates were immunoblotted using relevant antibodies. (C) A375 and A375R cells with indicated concentrations of PLX4032 or ATRA (20 μM), alone or in combination, for 24 h, after which cell viability was measured via MTT assays. Points, mean of three experiments; bars, S.D.; **p<0.05. (D) A375 and A375R cells with PLX4032 (1 μM) or the indicated concentrations of ATRA, alone or in combination, for 24 h, then the cells were harvested and whole-cell lysates were immunoblotted using appropriate antibodies. (E-F) A375 and A375R cells were exposed to PLX4032 (1 μM) or ATRA (20 μM), alone or in combination, in soft agar matrix and incubated at 37°C in a 5% CO2 atmosphere for 14 days. Colonies from three separate experiments are illustrated. The average number of colonies was measured. Columns, mean of three experiments; bars, S.D.; **p<0.01.
Discussion
A large proportion of melanoma patients displays BRAF V600E mutation, an oncogenic variant of the BRAF gene (27). BRAF V600E mutation plays a critical role in the progression of melanoma, which makes it an important target for chemotherapy (28). PLX4032 was developed as a specific inhibitor of BRAF V600E kinase (29, 30). Despite an impressive response rate of 80% at treatment initiation, melanoma patients exhibit acquired resistance to PLX4032 shortly after the commencement of chemotherapy (13). Accumulating evidence suggests that up-regulation of EGFR plays an important role in the development of acquired resistance through activation of either the RAF/MEK/ERK or PI3K/AKT pathways (12, 31-33). Herein, we identified PIN1 as a key regulator of EGFR expression in PLX4032-resistant melanoma cells. We found that expression of PIN1 is up-regulated in PLX4032-resistant A375R cells compared to parental A375 cells. Silencing of PIN1 in resistant cells reduced the activity of the RAF1-MEK1/2-ERK1/2 pathway, and restored PLX4032 sensitivity through down-regulation of EGFR expression. Furthermore, we showed that PLX4032, when used in combination with ATRA, a PIN1 inhibitor, reduced cell viability, anchorage-independent growth, and induced apoptosis in A375R cells. Taken together, our study outlines the important role of PIN1 in the development of PLX4032 resistance.
PIN1 selectively binds to a subset of phosphorylated proteins at pSer/Thr-Pro motifs, and induces cis/trans isomerization around the proline residue (18). PIN1 expression is increased in a wide range of cancers, including melanoma (19). PIN1 has been reported to promote melanoma progression through several mechanisms such as promotion of PIN1-FOXM1 signaling, PI3K/AKT signaling, among others (31, 34, 35). However, its role in the development of PLX4032 resistance is not fully understood. In this study, we found that PIN1 is up-regulated in PLX4032-resistant melanoma cells compared to PLX4032-sensitive parental cells. Two key mechanisms for resistance to PLX4032 in melanoma include activation of the PI3K/AKT pathway, and/or reactivation of MAPK signaling. EGFR functions upstream of both of these pathways and, therefore, up-regulation of EGFR plays a crucial role in the development of resistance. However, the underlying mechanisms involved in regulation of EGFR expression in resistant melanoma cells are poorly understood. Microphthalmia-associated transcription factor (MITF) is reported to modulate therapeutic resistance to PLX4032 by regulating EGFR expression, but no small molecule inhibitors of MITF currently exist, suggesting that alternative targets have to be identified to intervene in EGFR expression in resistant melanoma (36). Previous studies have shown that PIN1 enhances transcriptional activity of AP-2α, a transcription factor, to increase the expression of HER2 receptor, a tyrosine kinase receptor closely related to EGFR, in breast cancer (21). In addition, PIN1 directly interacts with HER2 to increase protein stability (37). Since EGFR shares structural similarity with HER2, and given that AP-2α is also responsible for EGFR expression, we examined whether PIN1 can regulate EGFR expression (38). Herein, we found that overexpression of PIN1 in A375 cells increased EGFR expression, and attenuated the responses of A375 cells to PLX4032, whereas silencing of PIN1 in resistant cells decreased EGFR expression and increased sensitivity to PLX4032. Our findings are in agreement with a previous report which shows that inhibition of PIN1 using a selective and cell-permeable inhibitor compound 37 or siRNA exerts strong synergistic effects with PLX4032 in SKmel28 cells, a melanoma cell line with BRAF V600E mutation (39). Even though our study does not explain whether PIN1 regulates EGFR expression at the transcriptional or protein levels, the data shown here clearly indicate that PIN1 is a key regulator of EGFR expression in PLX4032-resistant melanoma cells.

Pro-apoptotic effect is enhanced by treatment with PLX4032 and ATRA in combination in A375R cells (A) A375 and A375R cells were treated with PLX4032 (1 μM) or various doses of ATRA (5, 10 and 20 μM), alone or in combination, for 24 h, after which the cells were harvested and whole-cell lysates were immunoblotted using relevant antibodies. (B-C) A375 and A375R cells were treated with PLX4032 (1 μM) or ATRA (20 μM), alone or in combination, respectively. After 24 h, cells were harvested and used for FACS analysis as described in materials and methods. Columns, mean of three experiments; bars, S.D.; **p<0.01. (D) A375 and A375R cells were treated with PLX4032 (1 μM) or ATRA (20 μM), alone or in combination, respectively, for 24 h, then DNA fragmentation was detected. FITC, fluorescein isothiocyanate; DAPI, 4’,6-diamidino-2-phenylindole.

In vivo combination of PLX4032 and ATRA in PLX4032-resistant melanoma xenograft model. BALB/c mice were injected subcutaneously with A375 or A375R cells, respectively. On day 5 postinjection, mice were injected intraperitoneally with 20 mg/kg PLX4032 or 10 mg/kg ATRA for 25 days, alone or in combination, and tumor growth was measured. Shown are representative images of tumors (A), tumor weight (B), and tumor volume (C). Data are the means±SD, n=10, **p<0.01.
Current strategies to overcome PLX4032 resistance in melanoma are focused on either intermittent treatment schedules, development of novel RAF/MEK inhibitors, or combination therapies with AKT/MEK inhibitors (40, 41). The MEK inhibitors trametinib and cobimetinib are approved by the US FDA for the treatment of advanced melanoma in combination with BRAF inhibitors Dabrafenib and PLX4032, respectively (42). A phase III clinical study showed that combination of PLX4032 and cobimetinib increased progression-free survival from 6.9 months for PLX4032 alone to 9.9 months with the combination of agents (42). Even though the combination of PLX4032 and cobimetinib increases overall survival, resistance eventually develops. Moreover, this combination showed higher toxicity, suggestion that additional chemotherapeutic approaches have to be developed to overcome resistance (42). In this study, we demonstrated that PIN1 may be a potential target to overcome PLX4032 resistance. Even though PIN1 appears to represent a potential therapeutic target, most of the currently available PIN1 inhibitors lack the desired specificity and potency, making them unsuitable for clinical use. Recently, ATRA was identified as an inhibitor of PIN1 (22). ATRA is a retinoid compound and is already approved for clinical use in the treatment of acute promyelocytic leukemia (38). The therapeutic target(s) of ATRA has remained elusive for a long time, but recently mechanism-based screening revealed that ATRA directly binds to the active site of PIN1, inhibits its function, and selectively induces protein degradation in cancer cells (22). In addition, ATRA has been shown to inhibit the invasiveness of melanoma cells by reducing the secretion of matrix proteolytic enzymes (43, 44). Given that ATRA is a potent inhibitor of PIN1, we treated A375R cells with a combination of ATRA and PLX4032 to study their effects on the recovery of PLX4032 resistance. In our study, ATRA reduced the levels of PIN1 and, when combined with PLX4032, reduced cell viability and anchorage-independent growth of A375R cells compared to PLX4032 alone. Since A375R cells can evade PLX4032-induced apoptosis, we examined whether ATRA can restore the apoptosis-inducing function of PLX4032 in A735R cells. The data showed that the combination of ATRA and PLX4032 induced apoptosis in A375R cells. Finally, in vivo data showed that co-treatment with ATRA and PLX4032 reduces the growth of tumors formed by A375R cells in BALB/c mice. Collectively, these results indicate PIN1 as an attractive therapeutic target, the inhibition of which may potentially downregulate the expression of EGFR, resulting in subsequent suppression of the RAS/RAF/MEK pathway. Hence, our study not only reveals a novel mechanism for the development of PLX4032 resistance in melanoma, but also provides a novel treatment strategy to overcome such resistance.
Acknowledgements
This work was supported by research funds from Chosun University (2015).
Footnotes
↵* These Authors contributed equally to this study.
Authors' Contributions
G.K., P.Y.B. and C.H.O. designed and performed experiments, analyzed and interpreted data, and wrote the manuscript. H.S.C. conceived the study, designed experiments, interpreted data, and wrote the manuscript. All Authors reviewed the manuscript.
Conflicts of Interest
The Authors declare no conflicts of interest.
- Received November 4, 2019.
- Revision received November 11, 2019.
- Accepted November 18, 2019.
- Copyright© 2019, International Institute of Anticancer Research (Dr. George J. Delinasios), All rights reserved





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}