


Abstract
Background/Aim: Induction of endoplasmic reticulum (ER) stress is a novel approach to cancer treatment. This study investigated the ability of the clinically feasible combination of the human immunodeficiency virus protease inhibitors lopinavir and ritonavir to induce ER stress killing urological cancer cells. Materials and Methods: Renal cancer cells (769-P, 786-O) and bladder cancer cells (UMUC-3, T-24) were used to investigate the ability of the combination to induce ER stress and its mechanism of action. Results: The combination inhibited the growth of both renal and bladder cancer cells synergistically by inducing ER stress. The combination-induced ER stress increased the expression of AMP-activated protein kinase and suppressed the mammalian target of rapamycin pathway. It also increased the expression of a tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) receptor and thereby sensitized the cancer cells to TRAIL. Conclusion: The combination of lopinavir and ritonavir acts against urological cancer cells by inducing ER stress synergistically.
- Lopinavir
- ritonavir
- endoplasmic reticulum stress
- AMP-activated protein kinase
- tumor necrosis factor-related apoptosis-inducing ligand
- renal cancer
- bladder cancer
To date there is no curative treatment for patients with advanced urological malignances. Drug repositioning has emerged as a novel strategy for finding new anticancer agents: commonly used drugs are screened for their anticancer activity and clinically applied for cancer treatment (1, 2). This strategy lowers the cost of developing new cancer treatments and provides patients with promising new treatments quickly.
Ritonavir is an inhibitor of human immunodeficiency virus (HIV) protease used for the treatment of acquired immunodeficiency syndrome (AIDS). It inhibits cytochrome P450 and P-glycoprotein and has been used as a booster activating other antiviral drugs in clinical settings (3, 4). Lopinavir is also an HIV protease inhibitor, and a lopinavir/ritonavir formulation has been approved for the treatment of AIDS (5, 6).
Inducing endoplasmic reticulum (ER) stress is an emerging anticancer strategy (7), and lopinavir is one of the most potent ER stress inducers (8). In the present study, we tested our hypothesis that ritonavir would enhance the activity of lopinavir and that the combination would kill renal cancer cells and bladder cancer cells by inducing ER stress synergistically. Furthermore, we evaluated the combination's ability to increase the expression of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) receptor and thereby sensitize the cancer cells to TRAIL.
Materials and Methods
Cell culture. Human renal cancer cells (769-P, 786-O) and human bladder cancer cells (UMUC-3, T-24) were purchased from the American Type Culture Collection (Rockville, MD, USA). The cells were cultured in either Roswell Park Memorial Institute Medium 1640 (769-P and 786-O cells), Minimum Essential Medium (UMUC-3 cells), or McCoy's 5A medium (T-24 cells) containing 10% fetal bovine serum and 1.0% penicillin/streptomycin (Invitrogen, Carlsbad, CA, USA) at 37°C under 5% CO2 in a humidified incubator.
Reagents and antibodies. Ritonavir purchased from Toronto Research Chemicals (North York, ON, Canada) and lopinavir purchased from Selleck (Houston, TX, USA) were dissolved in dimethyl sulfoxide. Cycloheximide purchased from Enzo Life Sciences (Farmingdale, NY, USA) was dissolved in distilled water. Human recombinant TRAIL purchased from R&D Systems (Minneapolis, MN, USA) was dissolved in sterile phosphate-buffered saline (PBS) containing 0.1% bovine serum albumin. These solutions were stored at −80°C or −20°C until use. Primary antibodies for western blotting were used against the following: survivin and death receptor 5 (DR5) (Santa Cruz Biotechnology, Santa Cruz, CA, USA); cleaved poly(ADP-ribose) polymerase (PARP), S6 ribosomal protein, phosphorylated S6, eukaryotic translation initiation factor 4E-binding protein 1 (4EBP1), and endoplasmic reticulum resident protein 44 (ERp44 (Cell Signaling Technology, Danvers, MA, USA); glucose-regulated protein 78 (GRP78) and AMP-activated protein kinase (AMPK) (Proteintech, Rosemont, IL, USA); phorbol-12-myristate-13-acetate-induced protein 1 (NOXA; Abcam, Cambridge, UK), and actin (Millipore, Billerica, MA, USA). Horseradish peroxidase conjugated goat anti-mouse and goat anti-rabbit antibodies were purchased from GE Healthcare (Wauwatosa, WI, USA).
Assessing the combined effect of lopinavir and ritonavir on cell viability and colony formation. Cells were placed in 96-well culture plates (5×103 cells/well) 1 day before being treated for 48 h with 5-40 μM lopinavir with/without 5-40 μM ritonavir. Cell viability was then determined by MTS assay (n=6). For colony formation assay, cells were placed in 6-well plates (300-500 cells/well) 1 day before treatment and cultured for 48 h with lopinavir (40 μM for 769-P cells and 20 μM for T-24 cells) with/without ritonavir (10 μM for 769-P cells and 40 μM for T-24 cells). They were then given fresh media and allowed to grow for 1-2 weeks, after which they were then fixed with 100% methanol and stained with Giemsa's solution. The colonies were quantified by using the ImageJ-plugin ColonyArea (9) (n=3).
Evaluating apoptosis induction by the lopinavir-ritonavir combination. Cells were placed in 12-well culture plates (1.0×105 cells/well) 1 day before being cultured for 48 h with lopinavir (40 μM for 769-P and 786-O cells; 20 μM for UMUC-3 and T-24 cells) with/without ritonavir (10 μM for 769-P and 786-O cells; 40 μM for UMUC-3 and T-24 cells). Apoptotic cells were detected by annexin-V assay. To evaluate the expression of apoptosis-associated proteins, cells were treated for 48 h with lopinavir (20-40 μM for 769-P and 786-O cells; 10-20 μM for UMUC-3 and T-24 cells) with/without ritonavir (10 μM for 769-P and 786-O cells; 40 μM for UMUC-3 and T-24 cells). Changes in the expression of cleaved PARP, NOXA, and survivin were assessed by western blotting.
Evaluating ER stress induction and mammalian target of rapamycin (mTOR) suppression by the lopinavir-ritonavir combination. Cells were treated for 48 h with lopinavir (20-40 μM for 769-P and 786-O cells; 10-20 μM for UMUC-3 and T-24 cells) with/without ritonavir (10 μM for 769-P and 786-O cells; 40 μM for UMUC-3 and T-24 cells). Changes in the expression of GRP78, ERp44, AMPK, phosphorylated S6, S6, and 4EBP1 were assessed by western blotting.
Evaluating changes in the expression of TRAIL receptor by the lopinavir-ritonavir combination. Cells were treated with the combination (40 μM lopinavir and 10 μM ritonavir for 769-P cells; 20 μM lopinavir and 40 μM ritonavir for UMUC-3 cells) for 12, 24, and 48 h. Changes in the expression of the TRAIL receptor DR5 were evaluated by western blotting.
Evaluating sensitization of the cancer cells to TRAIL by the lopinavir-ritonavir combination. One day before treatment, cells for viability assay were placed in 96-well culture plates (5.0×103 cells/well) and cells for apoptosis assay were placed in 12-well culture plates (1.0×105 cells/well). The cells were then treated with the combination (40 μM lopinavir and 10 μM ritonavir for 769-P cells; 20 μM lopinavir and 40 μM ritonavir for UMUC-3 cells) for 24 h before being cultured with 25 ng/ml TRAIL in addition for another 24 h. Cell viability was measured by MTS assay (n=12) and apoptosis was evaluated by annexin-V assay.
Combination indices (CIs) for the combination of lopinavir and ritonavir against renal cancer cells and bladder cancer cells. CI <1,=1, and >1 indicates synergism, additive effect, and antagonism.
Evaluating cycloheximide's effect on cytotoxicity and apoptosis induction by the lopinavir-ritonavir combination. One day before treatment, cells for viability assay were placed in 96-well culture plates (5.0×103 cells/well) and cells for apoptosis assay were placed in 12-well culture plates (1.0×105 cells/well). The cells were then treated with the combination (40 μM lopinavir and 10 μM ritonavir for 769-P and 786-O cells; 20 μM lopinavir and 40 μM ritonavir for UMUC-3 and T-24 cells) with or without 5 μg/ml cycloheximide for 48 h. Cell viability was measured by MTS assay (n=12) and apoptosis was evaluated by annexin-V assay.
Evaluating cycloheximide's effect on ER stress induction, mTOR suppression, and increased DR5 expression by the lopinavir-ritonavir combination. Cells were treated for 48 h with the combination therapy (40 μM lopinavir and 10 μM ritonavir for 769-P and 786-O cells; 20 μM lopinavir and 40 μM ritonavir for UMUC-3 and T-24 cells) with or without 5 μg/ml cycloheximide. Changes in the expression of GRP78, ERp44, AMPK, and DR5 were assessed by western blotting.
Cell viability assay. Cell viability was determined by MTS assay (CellTiter 96 Aqueous kit; Promega, Madison, WI, USA) following the manufacturer's protocol. Briefly, the medium was replaced with 20 μl MTS solution in 100 μl fresh medium after treatment. The plates were then incubated for 30-60 min and read in a microplate autoreader.
Annexin-V assay. Treated cells were washed with PBS, harvested by trypsinization, and stained with annexin V and 7-amino-actinomycin D (7-AAD) (Beckman Coulter, Marseille, France) according to the manufacturer's instructions. The cells were analyzed by using a flow cytometer and CellQuest Pro Software (BD Biosciences, San Jose, CA, USA). A total of 10,000 cells were counted. Data were obtained from three independent experiments.
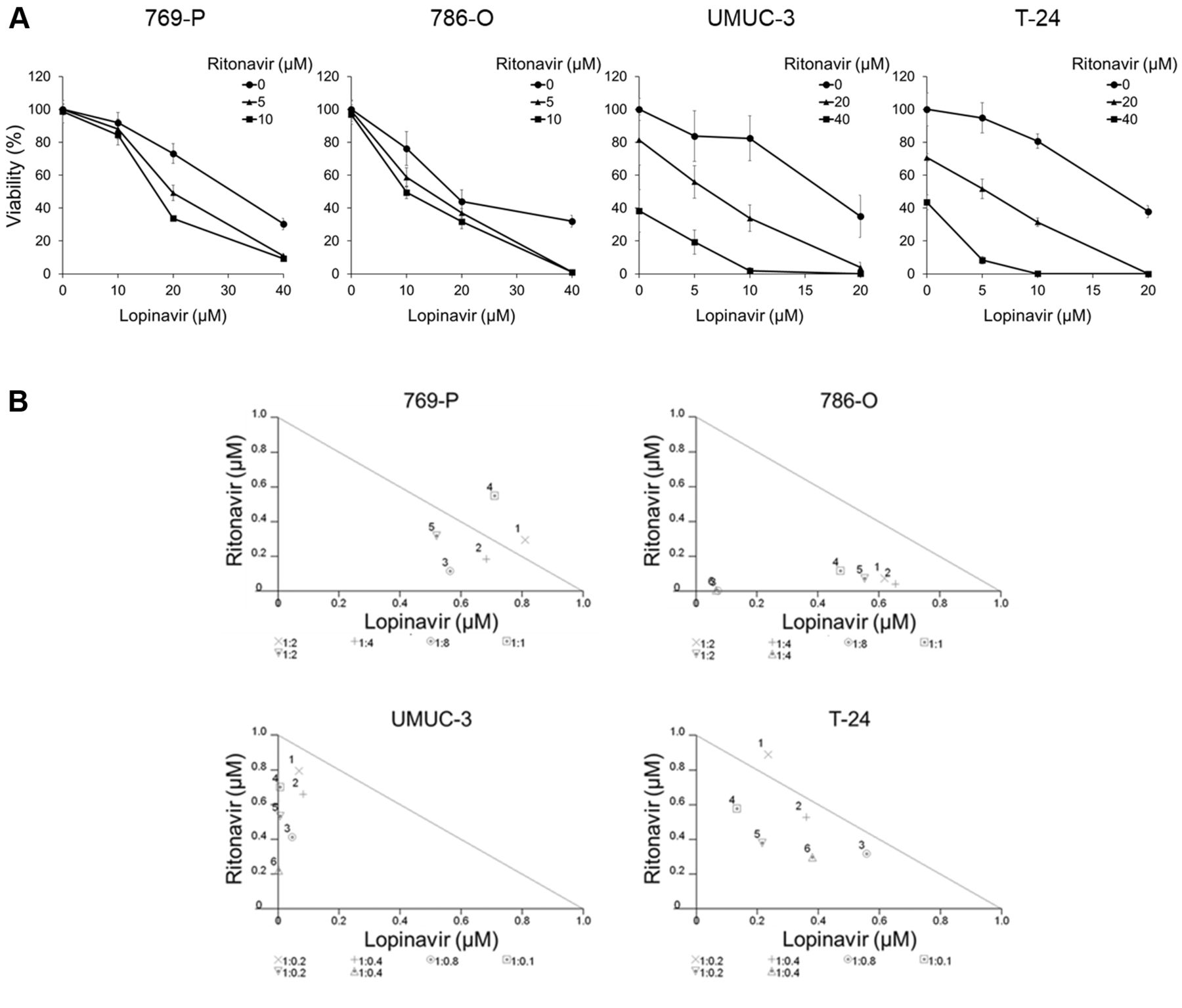


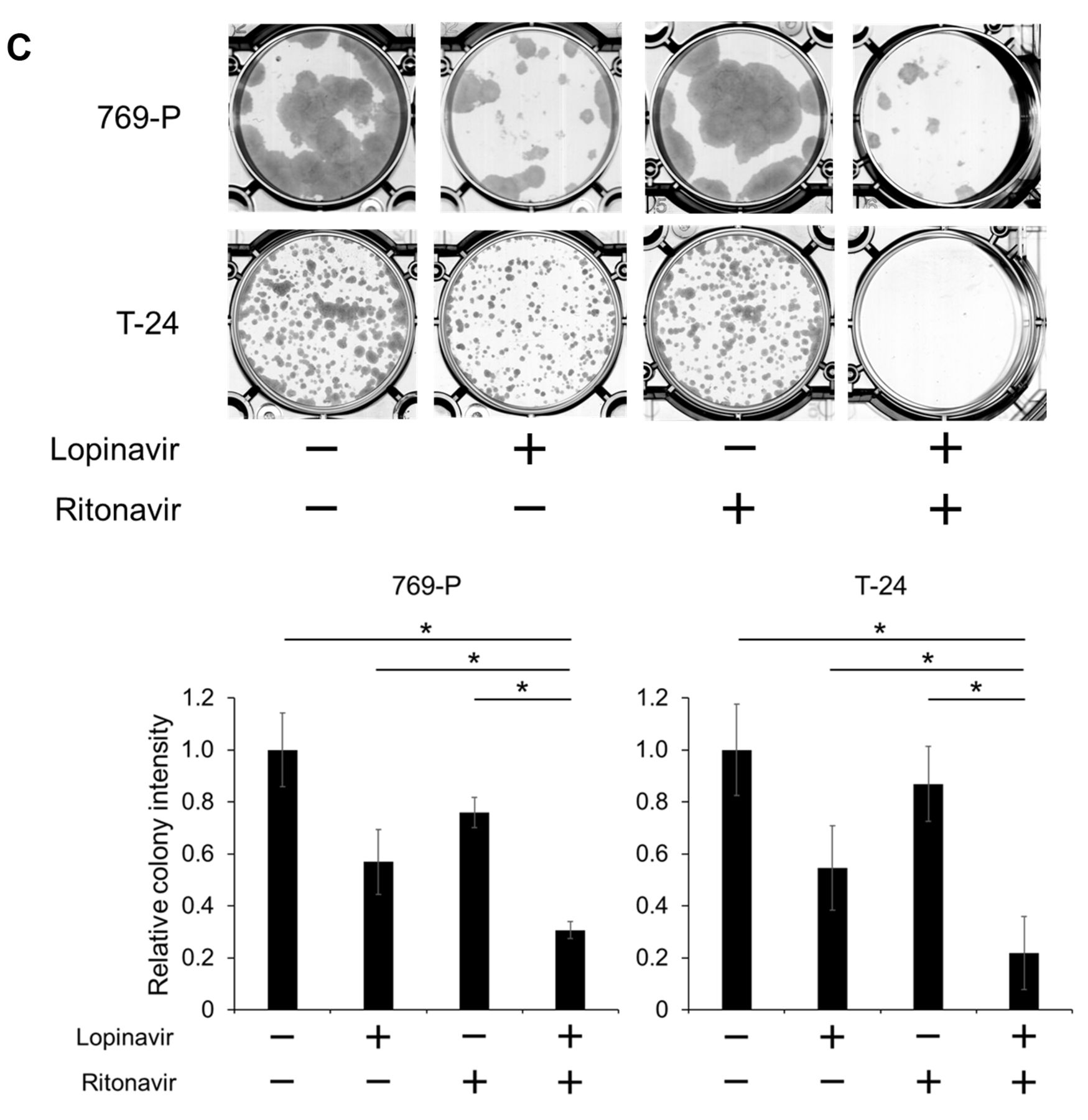
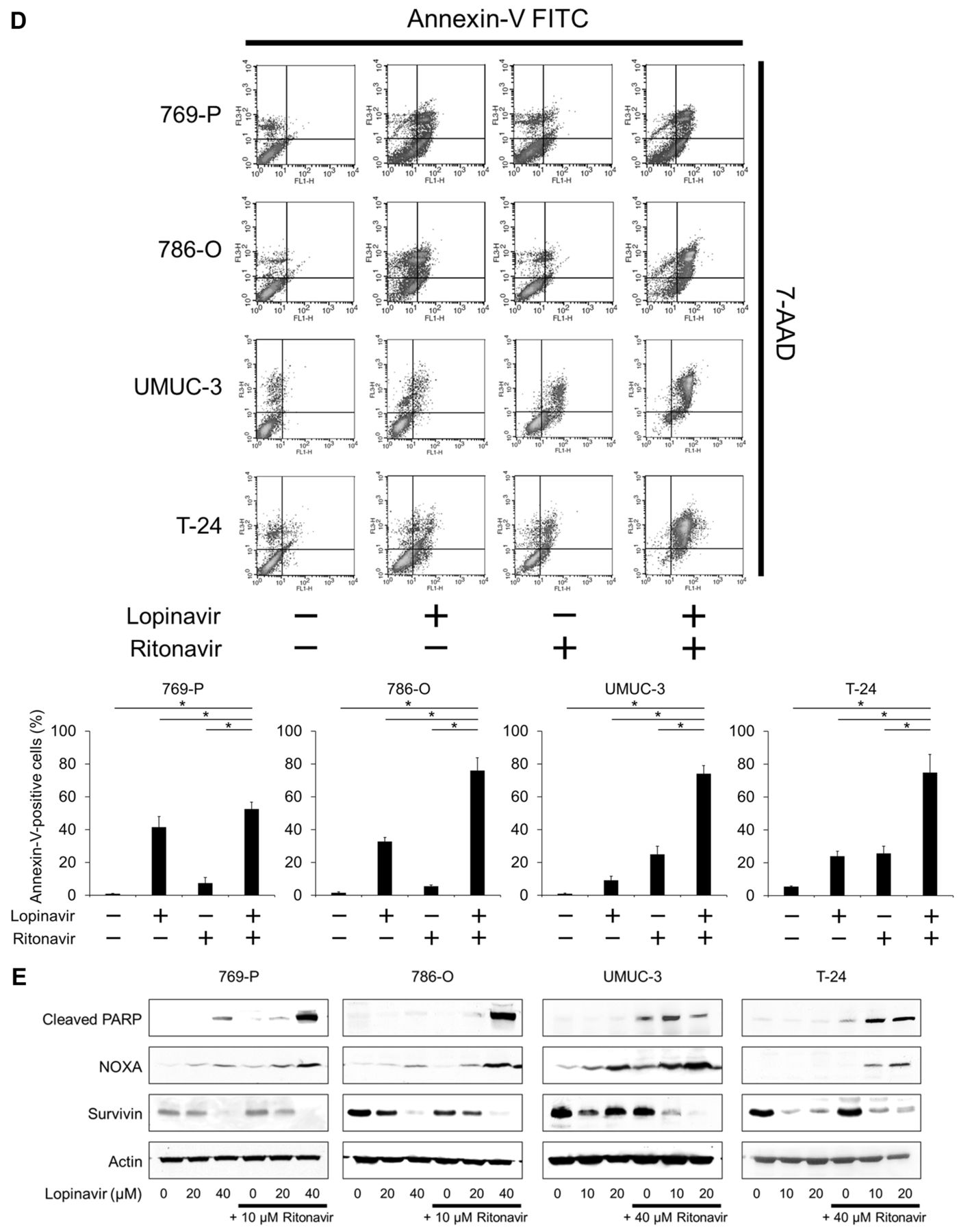
Co-administration of lopinavir and ritonavir synergistically inhibited the growth of cancer cells. A: Cell viability assay. Cells were treated for 48 h with 5-40 μM lopinavir with/without 5-40 μM ritonavir, and cell viability was measured using MTS assay. Data are the mean±SD, n=6. B: Isobologram analysis for the combination of lopinavir and ritonavir. C: Colony formation assay. Cells were treated for 48 h with lopinavir (40 μM for 769-P cells and 20 μM for T-24 cells) and/or ritonavir (10 μM for 769-P cells and 40 μM for T-24 cells). The cells were then given fresh media and allowed to grow for 1-2 weeks. Bar graphs show the relative colony intensity. Data are the mean±SD, n=3. D: Annexin-V assay. Cells were treated for 48 h with lopinavir (40 μM for 769-P and 786-O cells; 20 μM for UMUC-3 and T-24 cells) with/without ritonavir (10 μM for 769-P and 786-O cells; 40 μM for UMUC-3 and T-24 cells). Apoptotic cells were detected by annexin-V assay using flow cytometry; 10,000 cells were counted. Data are the mean±SD from three independent experiments. E: Western blotting for cleaved poly(ADP-ribose) polymerase (PARP), phorbol-12-myristate-13-acetate-induced protein 1 (NOXA), and survivin. Cells were treated for 48 h with 10-40 μM lopinavir with/without 10-40 μM ritonavir. Actin was used for the loading control. Representative blots are shown. FITC: Fluorescein isothiocyanate; 7-AAD: 7-amino-actinomycin D. *Significantly different at p=0.0495.
Western blotting. Treated cells were washed with PBS and whole cell lysates were obtained using radioimmunoprecipitation assay buffer. Equal amounts of proteins were separated by 12.5% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes. The membranes were blocked by 5% skimmed milk, and they were incubated overnight with the primary antibodies described above. Then the protein was detected by reaction with secondary antibodies and staining with chemiluminescence solution with the ECL Plus system (GE Healthcare).
Statistical analysis. CalcuSyn software (Biosoft, Cambridge, UK) was used for calculating the combination indexes according to the method by Chou and Talalay (10). Statistical significance of observed differences between samples was evaluated using the Mann-Whitney U-test (JMP Pro14 software; SAS Institute, Cary, NC, USA), and values of p<0.05 were considered to indicate a statistically significant difference.
Results
Co-administration of lopinavir and ritonavir synergistically inhibited the growth of cancer cells. According to the cell viability assay, the combination of lopinavir and ritonavir cooperatively inhibited the growth of the cancer cells (Figure 1A). The combined effect was synergistic under many of the treatment conditions (Figure 1B and Table I). Furthermore, the combination significantly inhibited the clonogenicity of the cancer cells (Figure 1C).
We next evaluated apoptosis induced by the combination. The combination increased the number of annexin-V positive cells significantly (Figure 1D), increasing the expression of cleaved PARP and NOXA, and reducing the expression of the anti-apoptotic protein survivin (Figure 1E). Thus, the combination of lopinavir and ritonavir was shown to induce apoptosis.
The combination induced ER stress and increased the expression of AMPK, thereby suppressing the mTOR pathway. We then evaluated the changes in the expression of the ER stress markers GRP78 and ERp44 to see whether the combination induced ER stress. Lopinavir itself induced no or only moderate ER stress, and ritonavir enhanced this stress prominently (Figure 2A).
Previous studies demonstrated that ER stress induced AMPK expression in malignancies (11, 12). Furthermore, AMPK is known to act against cancer by suppressing the mTOR pathway (13). As expected, the combination indeed increased the expression of AMPK synergistically, inhibited the expression of 4EBP1, and also inhibited S6 phosphorylation (Figure 2B), suggesting that the combination actually inhibited the mTOR pathway by increasing the expression of AMPK.

The combination of lopinavir and ritonavir induced endoplasmic reticulum (ER) stress and increased the expression of AMP-activated protein kinase (AMPK), thereby suppressing the mammalian target of rapamycin pathway. A: Western blotting for ER stress markers glucose-regulated protein 78 (GRP78) and endoplasmic reticulum resident protein 44 (ERp44). Cells were treated for 48 h with 10-40 μM lopinavir with/without 10-40 μM ritonavir. Actin was used for the loading control. Representative blots are shown. B: Western blotting for AMPK, phosphorylated S6 ribosomal protein (phosphorylated S6), S6, and eukaryotic translation initiation factor 4E-binding protein 1 (4EBP1). Cells were treated for 48 h with 10-40 μM lopinavir with/without 10-40 μM ritonavir. Actin was used for the loading control. Representative blots are shown.
The combination increased the expression of a TRAIL receptor and thereby sensitized cancer cells to TRAIL. ER stress induction reportedly increases the expression of TRAIL receptors (11, 14, 15). Therefore, we next evaluated whether the combination increases the expression of a TRAIL receptor and thereby sensitizes the cancer cells to TRAIL. The combination induced ER stress and increased the expression of the TRAIL receptor DR5, with the expression being highest after 24 h of treatment (Figure 3A). As expected, the combination sensitized the cancer cells to TRAIL: The ability of TRAIL to exert cytotoxicity and induce apoptosis was significantly enhanced when the cells were pretreated with the combination for 24 h (Figures 3B and C).
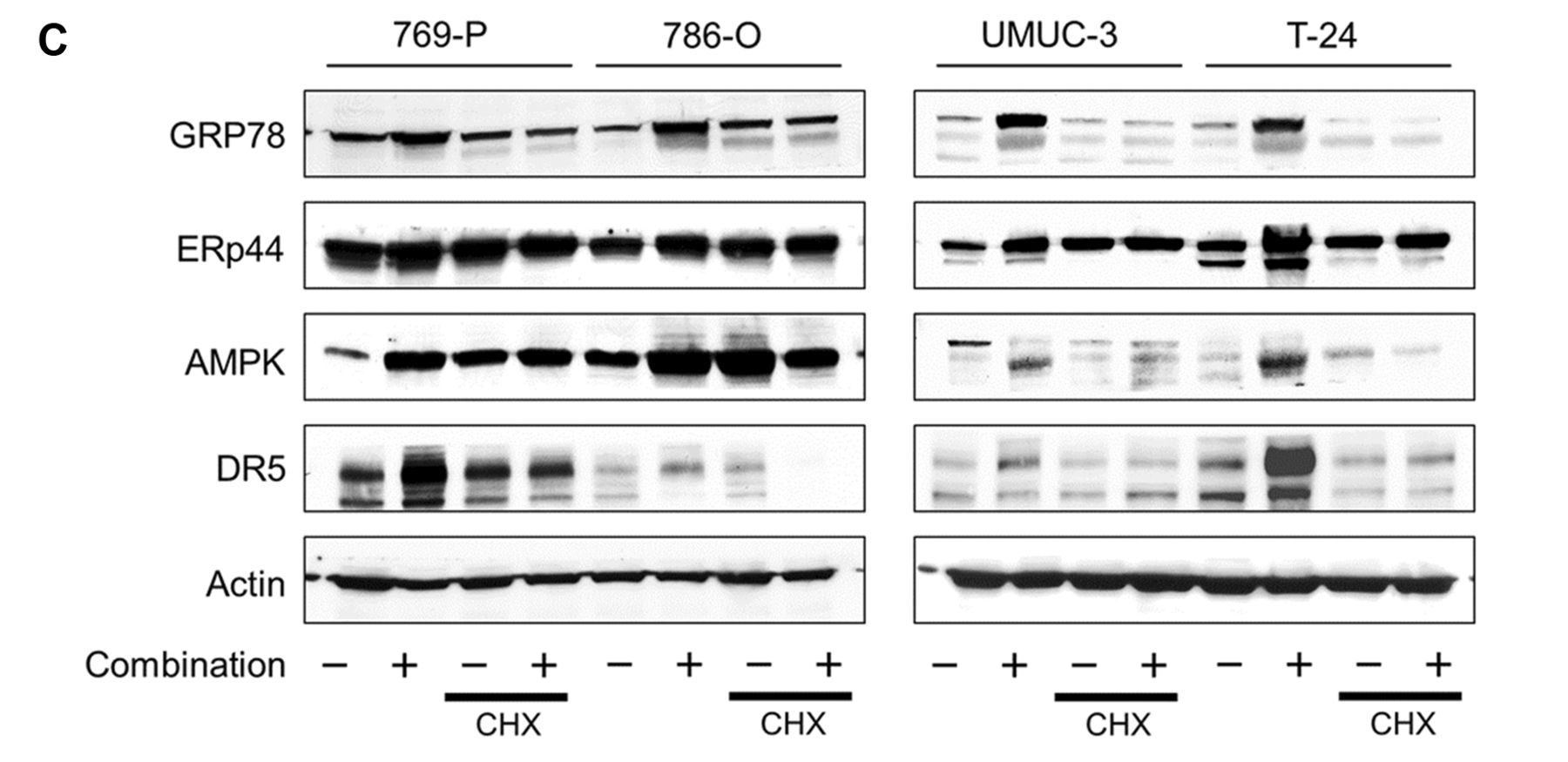
ER stress induction played a pivotal role in the anticancer effect of the combination. We then evaluated the role of ER stress induction in the action of combination of lopinavir and ritonavir. The ER stress inhibitor cycloheximide (16) attenuated the cytotoxicity of the combination of lopinavir and ritonavir and impaired its ability to induce apoptosis (Figures 4A and B). Cycloheximide inhibited the combination-increased expression of GRP78 and ERp44, confirming that it indeed suppressed ER stress induction (Figure 4C). Cycloheximide also reduced the combination-increased expression of AMPK and DR5. We inferred from cycloheximide effects that ER stress induction played a pivotal role in the combination's anticancer action.
Discussion
Inducing ER stress is an innovative approach to cancer therapy (7). HIV protease inhibitors are known to induce ER stress (4, 8, 14, 17, 18), and in the present study we demonstrated that the lopinavir–ritonavir combination synergistically suppressed the growth of both renal cancer cells and bladder cancer cells by inducing ER stress.
Because ritonavir has been shown not only to increase the amount of unfolded proteins by inhibiting molecular chaperones (4) but also to act as a chemical booster (3, 4), it is thought to be an ideal drug for inducing extensive ER stress in combination with other ER-stress-inducing agents (4, 19-21). Lopinavir, on the other hand, is also an HIV protease inhibitor and known as a strong ER stress inducer (8). We therefore believed co-administration of lopinavir and ritonavir would induce ER stress synergistically and thereby kill cancer cells effectively.

The combination of lopinavir and ritonavir increased the expression of a tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) receptor and thereby sensitized cancer cells to TRAIL. A: Western blotting for glucose-regulated protein 78 (GRP78) and death receptor 5 (DR5). Cells were treated for 12-48 h with the combination of lopinavir and ritonavir. Actin was used for the loading control. Representative blots are shown. Combination, 40 μM lopinavir and 10 μM ritonavir for 769-P cells; 20 μM lopinavir and 40 μM ritonavir for UMUC-3 cells. B: MTS assay. Cells were cultured for 24 h in medium with or without the combination of lopinavir and ritonavir (40 μM and 10 μM, respectively for 769-P cells; 20 μM and 40 μM, respectively for UMUC-3 cells). Then they were given 25 ng/ml TRAIL and incubated for another 24 h. Bar graphs show the relative viability (the viability of the control cells and that of the cells treated with the combination alone were both set at 1). Data are the mean±SD, n=12. *Significantly different at p=0.0001. C: Annexin-V assay. Cells were cultured for 24 h in medium with or without the combination of lopinavir and ritonavir. Then they were given 25 ng/ml TRAIL and incubated for another 24 h. Apoptotic cells were detected by annexin-V assay using flow cytometry; 10,000 cells were counted. Bar graphs show the increase in annexin-V positive cells by the addition of TRAIL. Data are expressed as mean±SD from three independent experiments. FITC: Fluorescein isothiocyanate; 7-AAD: 7-amino-actinomycin D. *Significantly different at p=0.0495.


Endoplasmic reticulum (ER) stress induction played a pivotal role in the anticancer effect of the lopinavir-ritonavir combination. A: MTS assay. Cells were treated with the combination of lopinavir and ritonavir (40 μM and 10 μM, respectively for 769-P and 786-O cells; 20 μM and 40 μM, respectively for UMUC-3 and T-24 cells) with or without 5 μg/ml cycloheximide (CHX) for 48 h. Bar graphs show the relative viability (the viability of the control cells and that of the cells treated with CHX alone were both set at 1). Data are the mean±SD, n=12. Significantly different at *p=0.0001. B: Annexin-V assay. Cells were treated with the combination of lopinavir and ritonavir with or without 5 μg/ml CHX for 48 h. Apoptotic cells were detected by annexin-V assay using flow cytometry; 10,000 cells were counted. Bar graphs show the combination-increased annexin-V positive cells. Data are the mean±SD from three independent experiments. FITC: Fluorescein isothiocyanate; 7-AAD: 7-amino-actinomycin D. Significantly different at *p=0.0495. C: Western blotting for glucose-regulated protein 78 (GRP78), endoplasmic reticulum resident protein 44 (ERp44), AMP-activated protein kinase (AMPK), and death receptor 5 (DR5). Cells were treated with the combination of lopinavir and ritonavir with or without 5 μg/ml CHX for 48 h. Actin was used for the loading control. Representative blots are shown.
As postulated, the combination of lopinavir and ritonavir induced ER stress cooperatively. In addition, we found that the combination increased the expression of the mTOR inhibitor AMPK and inhibited the mTOR pathway. AMPK is an energy sensor activated by ATP depletion to promote cellular catabolism of nutrients (22). Recently, it has also been shown to control cancer cell growth by suppressing the mTOR pathway (13, 22, 23). The mTOR pathway has been reported to play a key role in cell proliferation in both renal cancer and bladder cancer (24-26), therefore increasing AMPK expression would be a promising approach to killing those cancer cells. In the present study, we showed that AMPK expression in renal cancer cells and bladder cancer cells is increased by ER stress induced by the combination. ER stress induction can also trigger AMPK activation by decreasing cellular ATP levels (27, 28). Another important mechanism of AMPK activation is regulation of the AMPK-activating calcium/calmodulin-dependent kinase kinase (CaMKK)-beta by ER stress (12, 29, 30). Furthermore, our previous studies revealed that ER stress induction is closely associated with AMPK activation (31, 32). Thus, ER stress is closely related to regulation of AMPK.
TRAIL, a member of the tissue necrosis factor family, induces apoptosis when it binds to TRAIL receptors (33, 34). Some studies reported that higher levels of intrinsic TRAIL expression are associated with more favorable response to surgical treatment and higher survival rates (35-37); increasing the susceptibility of cancer cells to TRAIL is thought to be an attractive approach to cancer therapy. Interestingly, we found that the combination of lopinavir and ritonavir triggered TRAIL-receptor–mediated cytotoxicity and apoptosis by increasing the expression of the TRAIL receptor DR5. Several studies have shown that ER stress induction increased the expression of TRAIL receptors, thereby sensitizing cancer cells to TRAIL and inducing TRAIL-mediated apoptosis (11, 14, 38). Compatible with those studies, the increased DR5 expression we found was shown to be due to ER stress induced by the combination.
Because the lopinavir–ritonavir combination has widely been used for HIV treatment, it would be relatively easy to reposition it for other indications and use it to treat renal cancer and bladder cancer. Furthermore, the combination might be effective in killing cancer cells irrespective of cancer type because induction of ER stress is associated with the ubiquitin-proteasome pathway, a protein homeostasis mechanism essential for cell survival (7, 39). In fact, the combination effectively killed completely different types of cancer: renal cancer and bladder cancer. Its efficacy should therefore also be tested in other types of cancer.
To our knowledge, this is the first study showing that the lopinavir–ritonavir combination killed urological cancer cells by inducing ER stress and thereby increasing the expression of AMPK and DR5. The present study provides a framework for applying the combination to cancer treatment.
Footnotes
Authors' Contributions
K.O., M.I., and A.S. designed the study. K.O. carried out all the experiments. K.O., M.I., T.A., and A.S. contributed to the interpretation of the results. K.O. wrote the article. A.S. supervised the study and edited the article. All Authors read and approved the final article.
This article is freely accessible online.
Conflicts of Interest
None.
- Received August 30, 2019.
- Revision received October 2, 2019.
- Accepted October 4, 2019.
- Copyright© 2019, International Institute of Anticancer Research (Dr. George J. Delinasios), All rights reserved





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}