


Abstract
Background/Aim: Multidrug resistance leads to therapeutic difficulties. There is great interest in experimental chemotherapy regarding multidrug resistance inhibitors and new anticancer agents. The aim of this study was to evaluate the anticancer activity of exocyclic sulfur and selenoorganic compounds on mouse T-lymphoma cell lines. Materials and Methods: A series of eighteen sulfur and selenium analogues of 2[1H]-pyrimidinone and hydantoin derivatives were evaluated towards their efflux modulating, cytotoxic and antiproliferative effects in mouse T-lymphoma cells. The combination assay with doxorubicin on multidrug resistant mouse T-lymphoma cells was performed in order to see the nature of drug interactions. Crystal structures were determined for two selected compounds with the highest efflux-modulating activity. Results: The sulfur analogues with aromatic rings almost perpendicular to pyrimidinethione ring at positions 1 and 6 showed the highest efflux inhibitory action, while all selenium analogues showed good antiproliferative and cytotoxic activities. Conclusion: The sulfur analogues can be modified towards improving their efflux inhibitory activity, whereas the selenium towards antiproliferative and cytotoxic activities.
- Pyrimidinethione
- pyrimidineselenone
- thiohydantoin
- selenohydantoin
- crystal structure
- P-gp inhibitor
- antiproliferative activity
- combination assay with doxorubicin
Cancer is still a major human health problem leading very often to mortality. According to WHO data, cancer as the second leading cause of death, was responsible for approximately 14 million new cases in the year 2012 and killed 8.8 million people in 2015 (1). There is still a need to search for new anticancer drugs, due to the increasing multidrug resistance leading to therapeutic difficulties.
There is great interest in experimental chemotherapy regarding multidrug resistance (MDR) inhibitors. The cellular overproduction of MDR efflux pumps for example ABCB1 (P-glycoprotein, P-gp) is one of the more relevant mechanisms underlying MDR, which is capable to promote the efflux of cytotoxic drugs out of the cells (2). ABCB1 belongs to the ATP-binding cassette (ABC) transporter proteins, involved in the transport of various substrates e.g. cholesterol, drugs, chloride ion, toxins, across membranes (3). In cancer cells, the overexpression of ABCB1 is responsible for reduction of the intracellular concentration of the anticancer drugs, which in consequence leads to treatment failure (4). It should be pointed out that several clinical trials targeting ABCB1 with small molecules, first-generation (verapamil, cyclosporine A, quinidine) (5, 6), second-generation (dexverapamil, cinchonine, valspodar) (7, 8) and third-generation (zosuquidar, tariquidar, laniquidar) inhibitors have failed due to their toxicity (9, 10). Finding non-toxic inhibitors, capable of decreasing the ABCB1-mediated drug efflux, is a promising strategy for improving the effectiveness of cancer treatment.
Recently, a number of different compounds were designed as potential MDR modulators, including new chemical groups of ABCB1 inhibitors, i.e. derivatives of xanthone (11), benzoxanthone (12), hydantoin and thiohydantoin (13, 14), benzodipyranone (15), indol alkaloid (16), selenoesters (17, 18), epigallocatechin and dihydromyricetin (19).
Taking this into consideration, we decided to evaluate eighteen sulfur and selenium analogues of 2[1H]-pyrimidinone (1-8) and hydantoin (9) derivatives (Figure 1) towards their efflux modulating, cytotoxic and antiproliferative effects in sensitive and resistant mouse T-lymphoma cells. Furthermore, the selected compounds were investigated in combination with the anticancer drug doxorubicin. The aim of our study was to compare the biological activity of sulfur and selenium compounds and to analyze crystal and molecular structures in the search of a structural feature that can be responsible for their activity.
The investigated compounds were synthesized according to the procedures published earlier (20, 21). In this paper, two new crystal structures of selected 2[1H]-pyrimidinethione derivatives are reported: 4-methyl-1-(4-methylphenyl)-6-phenyl-2[1H]-pyrimidinethione (3S) and 4-methyl-1-(4-methoxyphenyl)-6-phenyl-2[1H]-pyrimidinethione (4S). These compounds differ in the substituent at the aromatic ring and show the best activity towards inhibition of ABCB1, wherein the activity of 4S is 3-fold higher than that of 3S. The arrangement of rings may be related to the biological activity. In order to see mutual orientation of all three rings, a deeper insight into the structural properties of the presented compounds was performed with the use of X-ray diffraction analysis. Moreover, we compared crystal structures of active compounds with the known crystal structure of the not active compound 1S (22). We present the results of the biological studies of sulfur (1S-9S) and selenium (1Se-9Se) analogues of pyrimidinone and hydantoin derivatives (Figure 1) in order to see how the change of the sulfur atom to selenium can influence the ABCB1 inhibition, cytotoxic and antiproliferative activities.
Materials and Methods
Crystal structures determination. Crystals suitable for X-ray structure analysis were obtained from ethanol solution by slow evaporation of the solvent at room temperature. The intensity data were collected on the Bruker-Nonius Kappa CCD four circle diffractometer equipped with a Mo (0.71069 Å) Kα radiation source. The phase problem was solved by direct methods using SHELXS and the structures were refined by SHELXL (23) programs. For molecular graphics MERCURY (24) program was used.
Compound 3S: C18H16N2S, Mr=292.39, crystal size=0.04×0.18×0.41 mm3, monoclinic, space group P21/c, a=10.9600(2) Å, b=25.4850(5) Å, c=12.9280(2) Å, β=121.028(1)°, V=3094.3(1) Å3, Z=8, T=100(2) K, 22,417 reflections collected, 7,059 unique reflections [Rint=0.0474], R1=0.0429, wR2=0.0870 [I>2sigma(I)].
Compound 4S: C18H16N2OS, Mr=308.39, crystal size=0.17×0.36×0.55 mm3, monoclinic, space group P21/c, a=12.6920(3) Å, b=8.9240(2) Å, c=16.8230(3) Å, β=125.283(2)°, V=1555.42(7) Å3, Z=4, T=100(2) K, 12217 reflections collected, 3556 unique reflections [Rint=0.0334], R1=0.0346, wR2=0.0849 [I>2sigma(I)].
CCDC 1833588 and 1833589 contain the supplementary crystallographic data for 3S and 4S, respectively. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
Biological assays
Cell lines. The L5178Y mouse T-lymphoma cells (PAR) (ECACC Cat. No. 87111908, obtained from FDA, Silver Spring, MD, USA) were transfected with pHa MDR1/A retrovirus (25). The ABCB1-expressing cell line L5178Y (MDR) was selected by culturing the infected cells with colchicine. L5178Y (parent) mouse T-cell lymphoma cells and the L5178Y human ABCB1 transfected subline were cultured in McCoy's 5A medium (Sigma-Aldrich, Steinheim, Germany) supplemented with 10% heat-inactivated horse serum (Sigma-Aldrich), 200 mM L-glutamine (Sigma-Aldrich) and a penicillin streptomycin (Sigma-Aldrich) mixture in concentrations of 100 U/l and 10 mg/l, respectively.
Rhodamine-123 (R123) accumulation assay by flow cytometry. The cell number of L5178Y MDR and L5178Y parental cell lines were adjusted to 2×106 cells/ml, re-suspended in serum-free McCoy's 5A medium and distributed in 0.5 ml aliquots into Eppendorf centrifuge tubes. The tested compounds were added at 2 and 20 μM concentrations and the samples were incubated for 10 min at room temperature. Verapamil (purity: ≥99%; Sigma-Aldrich) was applied as positive control. DMSO at 2% was also added as solvent control. Next, 10 μl (5.2 μM final concentration) of the fluorochrome and ABCB1 substrate R123 (Sigma-Aldrich) were added to the samples and the cells were incubated for further 20 min at 37°C, washed twice and re-suspended in 0.5 ml PBS for analysis. The results obtained from a representative flow cytometry experiment measuring 20,000 individual cells of the population were evaluated using Partec CyFlow® flow cytometer (Partec, Münster, Germany). The percentage of mean fluorescence intensity was calculated for the treated MDR cells as compared with the untreated cells. A fluorescence activity ratio (FAR) was calculated based on the following equation which relates to the measured fluorescence values:

Assay for cytotoxic and antiproliferative effect. The compounds were diluted in a volume of 100 μl medium. Then, 1×104 cells (for cytotoxic assay) and 6×103 (for antiproliferative assay) in 100 μl of medium were added to each well, with the exception of the medium control wells (17). The culture plates were incubated at 37°C for 24 and 72 h; at the end of the incubation period, 20 μl of MTT (thiazolyl blue tetrazolium bromide; Sigma-Aldrich) solution (from a 5 mg/ml stock) were added to each well. After incubation at 37°C for 4 h, 100 μl of SDS (sodium dodecyl sulfate; Sigma-Aldrich) solution (10% in 0.01 M HCI) were added to each well and the plates were further incubated at 37°C overnight. Cell growth was determined by measuring the optical density (OD) at 540 nm (ref. 630 nm) with Multiscan EX ELISA reader (Thermo Labsystems, Cheshire, WA, USA). Inhibition of the cell growth was determined according to the formula:

Checkerboard combination assay. The dilutions of doxorubicin (2 mg/ml, Teva Pharmaceuticals, Budapest, Hungary) were made in a horizontal direction in 100 μl, and the dilutions of the resistance modifiers vertically in the microtiter plate in 50 μl volume. The L5178Y MDR mouse T-lymphoma cells were re-suspended in culture medium and distributed into each well in 50 μl containing 6×103 cells, with the exception of the medium control wells, to a final volume of 200 μl per well. The plates were incubated for 72 h at 37°C in a CO2 incubator and at the end of the incubation period, the cell growth was determined by the MTT staining method, as earlier described. Drug interactions were evaluated using CompuSyn software. Combination index (CI) values at 50% of the growth inhibition dose (ED50), were determined using CompuSyn software (www.combosyn.com, ComboSyn, Inc., Paramus, NJ, USA) to plot four to five data points to each ratio. CI values were calculated by means of the median-effect equation, where CI<1, CI=1, and CI>1 represent synergism, additive effect (or no interaction), and antagonism, respectively.
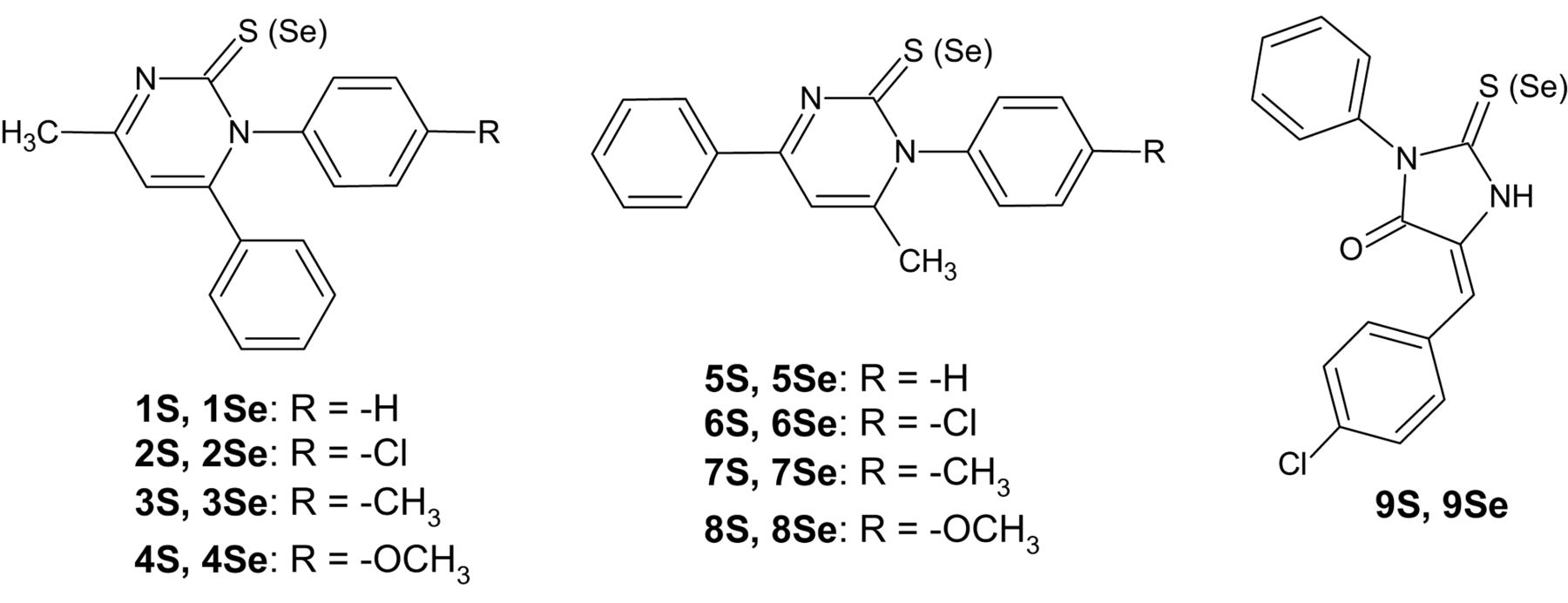
Chemical structures of investigated compounds.
Results
Crystal and molecular structures. The projections of molecular geometry in the crystal structures with the atom numbering are shown in Figure 2. The asymmetric unit of 3S consists of two independent molecules (one molecule is labeled as A, the second as B), while 4S one molecule.
The angle between the plane of 2[1H]-pyrimidinethione ring and the plane of aromatic ring at N1 atom is 67.02(6)°, 78.27(6)° and 82.82(5)°, whereas between the plane of 2[1H]-pyrimidinethione ring and aromatic ring at C6 is 56.14(7)°, 64.24(6)° and 76.16(5)° for 3S molecule A, 3S molecule B and 4S, respectively. The angle between the planes of two aromatic rings is 59.95(6)°, 66.27(6)° and 62.27(5)° for 3S molecule A, 3S molecule B and 4S, respectively.
The selected torsion angles, observed in the crystal structures of 1,6-diphenyl-2[1H]-pyrimidinethione derivatives presented in this paper and determined earlier (22), are listed in Table I. The comparison of them shows the slight differences of mutual orientations of aromatic rings and 2[1H]-pyrimidinethione ring.
The comparison of selected torsion angles [°] for 1,6-diphenyl-2[1H]-pyrimidinethione derivatives.
Nitrogen and sulfur atoms as acceptors and hydrogen atoms directly bonded to carbon atoms as donors, are engaged in weak intermolecular hydrogen bonds. Parameters of these interactions are listed in Table II. In the crystal structure of 3S carbon atoms of all three rings are engaged in intermolecular contacts, whereas in 4S only carbon atoms of aromatic rings and the methyl group at C4 atom. Substituents at aromatic ring are not engaged in any intermolecular interactions. There is an oxygen atom in the structure of 4S, which could be an acceptor in hydrogen bonds, but in the presented crystal structure is not involved in any intermolecular hydrogen bonds.
Efflux modulating effects. The efflux-modulating effects of 1S-9S and 1Se-9Se were investigated in the sensitive parental (PAR) and resistant (MDR) mouse T-lymphoma cells overexpressing ABCB1 using the standard R123 functional assay (17). The results are expressed as the fluorescence activity ratio (FAR) values, which represent the accumulation of R123 in L5178Y-MDR and L-5178Y-PAR cells (Figure 3). When FAR value is higher than one, the modulation of ABCB1 occurs, while FAR value that is higher than ten implicates strong modulation (26). The modulating effect was observed for almost all 1,6-diphenyl-2[1H]-pyrimidinethione/selenone derivatives (1-4), but only five compounds were more effective than the reference inhibitor verapamil and two of them (3S and 4S) showed strong modulation of ABCB1 at 20 μM. Other 1,4-diphenyl-2[1H]-pyrimidinethione/selenone derivatives (5-8) and thiohydantoin/selenohydantoin derivative (9) displayed lower activity than verapamil at 20 μM. The tested compounds had no efflux pump inhibiting effect at 2 μM.

The molecular structures of 3S (molecule A) and 4S in the crystalline state.
Parameters of weak intermolecular hydrogen bonds.
Cytotoxic and antiproliferative effects. All compounds were examined for their cytotoxic effect in sensitive (PAR) and resistant (MDR) mouse T-lymphoma cells (17). As shown in Table III, all selenium analogues displayed cytotoxicity in both cell lines. Almost all sulfur analogues with the exception of two (5S and 9S) showed no cytotoxic effect (IC50 >100 μM) in the mouse T-lymphoma cell lines.
The antiproliferative activity of the presented compounds was investigated in sensitive (PAR) and resistant (MDR) mouse T-lymphoma cells (Table III) (27). The tested selenium analogues (1Se-9Se) showed significant antiproliferative profiles with IC50 in the range 0.9-1.7 μM for 1Se-4Se, 2.0-4.5 μM for 5Se-9Se. The sulfur analogues displayed much weaker activity from 35 to over 100 μM except for 5S and 9S.
Checkerboard combination assay. For compounds with efficient antiproliferative activity (1Se-9Se, 5S and 9S), their ability to make MDR cells more susceptible to anticancer drug action was assessed using checkerboard combination assay. These compounds were tested together with the anticancer drug doxorubicin in order to get information about the nature of interactions between them (Table IV). In Table IV the best combination ratios of doxorubicin and tested compounds are presented. Combination index (CI) in the third column (Table IV) is based on the 50% of the growth inhibition dose (ED50). The nature of interactions is determined on the basis of the CI value as a synergism (CI<1), antagonism (CI>1) or additive effect (CI=1) (28).
Discussion
Previously, several classes of compounds have been described as cytotoxic agents, adjuvants in combination with doxorubicin, ABCB1 modulators and apoptosis inducers, such as phenothiazines (29), steroidal compounds (30) and hydantoin derivatives (31). The presented compounds showed different biological activities. The analyzed compounds contain two aromatic rings in three different locations: (i) 1-4 possess the closest phenyl substituents, in N1 and C6 positions of 2[1H]-pyrimidinethione/selenone ring; (ii) 5-8 have the most distant phenyl substituents, in N1 and C4 positions of 2[1H]-pyrimidinethione/selenone ring and (iii) 9 contains the distant phenyl substituent at N3 and benzylidene substituent at C5 of 2-thio/selenohydantoin ring (Figure 1). The positions of the aromatic rings and the presence of sulfur or selenium atom are connected with biological activities.
The results obtained for the modulation of ABCB1 mediated efflux showed the best activity for sulfur analogues with phenyl substituent at neighboring atoms (N1 and C6) defined as location (i). These compounds showed also no cytotoxic effects in both cell lines (PAR and MDR). A deeper insight into the structural properties of the 1,6-diphenylpyrimidinethione derivatives (1S, 3S, 4S) using the X-ray crystal structure analysis allowed us to determine the mutual orientation of the rings. Compound 4S with highest activity towards inhibition of ABCB1 showed the conformation with aromatic rings the closest to perpendicular to the plane of 2[1H]-pyrimidinethione ring. Other compounds possessed these rings more twisted. Compound 1S, containing unsubstituted aromatic rings, was not more active than verapamil, while introducing the substituent to aromatic ring at N1 atom increased this activity. The introduction of an electron-withdrawing substituent, namely chlorine atom (2S), led to a 7-fold increase in the efflux modulatory effect. However, an electron-donating substituent in aromatic ring, namely methyl group (3S), increased 11-fold the efflux modulatory effect, while methoxy group (4S), a stronger electron-donating substituent increased 30-fold ABCB1 inhibiting activity.

Rhodamine 123 retention in the presence of the compounds at 20 μM and verapamil at 20 μM as a positive control on multidrug-resistant T-lymphoma cells (MDR).
Interestingly, the replacement of sulfur atom with selenium atom increased almost 8-fold the activity of 1Se in comparison to 1S. The comparison of molecular geometries of these two compounds did not show significant differences (22). In this case the presence of selenium atom seems to be important for biological activity. However, the chlorine substituent in aromatic ring decreased the capacity of 2Se to inhibit the ABCB1 efflux pump, giving a value of FAR almost the same as for 1S. The introduction of a methyl substituent to the aromatic ring in 1Se (compound 3Se) improved activity in comparison to 2Se, while the presence of methoxy group in 4Se caused a lower activity than verapamil.
Cytotoxic activity among investigated compounds is mainly related to the presence of the selenium atom. The role of the aromatic rings' locations seems to be less important. All selenium analogues displayed high cytotoxic effects in both sensitive and resistant cell lines. Only two sulfur analogues (5S and 9S) showed cytotoxic activity. Sulfur analogues with modulating effect on ABCB1 (1S-4S) were not cytotoxic on the tested lines.
The capacity of the investigated compounds regarding antiproliferative activity is correlated with the cytotoxicity effect: all cytotoxic selenium analogues displayed antiproliferative activity with IC50 in the range of 0.9-6 μM in both cell lines (PAR and MDR). The sulfur analogues, excluding 5S and 9S, showed 10-fold weaker antiproliferative activity. The combination assay with doxorubicin on MDR cells for compounds with potent antiproliferative activity showed that selenium derivatives with (i) location of the rings (1Se-4Se) displayed antagonism, derivatives with (ii) location (5Se-8Se) moderate antagonism/synergism, while the third group (iii), namely derivative of 2-thio/selenohydantoin (9S, 9Se), synergism.
To summarize, the presented results allowed us to identify new compounds (1S-4S) showing potency to inhibit the cancer MDR efflux pump ABCB1. The introduction of an electron-donating substituent in the phenyl ring at N1 atom increased the activity. These results point out the 1,6-diphenyl-2[1H]-pyrimidinethione scaffold for further structural modifications in order to improve the efflux modulation on ABCB1. For the most active compound (4S) the mutual orientations of aromatic and pyrimidinethione rings show the closest angles to 90 degrees between the rings. All selenium analogues showed very potent cytotoxic and antiproliferative activities. Two derivates of the 1,4-diphenyl-2[1H]-pyrimidineselenone (5Se, 7Se) exerted a synergistic effect in combination with doxorubicin. Both 2-thiohydantoin and 2-selenohydantoin derivatives (9S, 9Se) improved the cytotoxicity of doxorubicin synergistically.
Cytotoxic and antiproliferative effects of the compounds on sensitive L5178Y parental (PAR) and resistant (MDR) mouse T-lymphoma cells.
Type of interaction between compounds and doxorubicin against multidrug-resistant (MDR) mouse T-lymphoma cells.
Acknowledgements
The Authors would like to thank Prof. Jadwiga Handzlik for her comprehensive discussion. The work was supported by Pedagogical University of Cracow (BS-465/G/2018).
- Received May 24, 2018.
- Revision received June 14, 2018.
- Accepted June 15, 2018.
- Copyright© 2018, International Institute of Anticancer Research (Dr. George J. Delinasios), All rights reserved
References
In this issue





{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.