


Abstract
Background/Aim: Eribulin mesylate has been approved for advanced or metastatic breast cancers subjected to at least two previous chemotherapy regimens. The present multicenter, phase II, single-arm study assessed the efficacy and safety of a first-line regimen of eribulin plus trastuzumab for untreated advanced or metastatic HER2-positive breast cancer. Patients and Methods: Enrolled patients received eribulin (1.4 mg/m2 intravenously; I.V.) on days 1 and 8 of each 21-day cycle, an initial trastuzumab dose (8 mg/kg I.V.) on day 1, and 6 mg/kg of trastuzumab on day 1 of each subsequent cycle. The primary endpoint was the response rate (RR). The secondary endpoints were progression-free survival (PFS), overall survival (OS), duration of response (DOR), and safety. Twenty-eight patients (median age: 62.5 years) received a median of 12 (range: 2–53) cycles of eribulin plus trastuzumab. Results: The RR was 53.6% [complete response (CR), 4; partial response (PR), 11] with a median PFS of 344 days. The clinical benefit rate was 64.0%. Grade 3/4 adverse events were observed in 12 (42.9%) patients. For details, neutropenia in 8 (28.6%) patients, peripheral neuropathy in 2 (7.1%) patients, interstitial pneumonia in 1 (3.6%) patient, ALT elevation in 1 (3.6%) patient, osteonecrosis of the jaw in 1 (3.6%) patient, and fatigue in 1 (3.6%) patient. The patient with osteonecrosis received denosumab, too. No symptomatic congestive heart failure was observed. Conclusion: Combination therapy of eribulin plus trastuzumab is acceptable in efficacy and safety, and a capable option for first-line advanced or recurrent HER2-positive breast cancer.
Breast cancer is the most common cancer among women, and the numbers of affected patients and related deaths continue to increase annually. In Japan, more than 90,000 patients are newly diagnosed with breast cancer each year, and the annual number of deaths due to breast cancer currently exceeds 13,000 (1). Currently, human epidermal growth factor receptor-2 (HER2)-positive tumors account for approximately 15% of all breast cancers, and HER2 overexpression has been identified as a poor prognosticator.
Currently, the molecular targeted drug trastuzumab is recommended as a standard therapy for HER2-positive metastatic breast cancer. However, the reported response rates following trastuzumab monotherapy was 19% (2). This response rate can be enhanced by combining trastuzumab with other chemotherapeutic drugs. The Physician Data Query (PDQ) (3) recommends that a standard first-line treatment for stage IIIB, IIIC, and IV progressive and relapse HER2-positive breast cancers should comprise single-agent chemotherapy + trastuzumab. The American Society of Clinical Oncology (ASCO) guideline (4) recommends a combination regimen comprising trastuzumab + pertuzumab + taxane for the first-line treatment of HER2-positive recurrent breast cancer. The National Comprehensive Cancer Network (NCCN) guidelines (5) recommend the administration of a preoperative trastuzumab-containing chemotherapy regimen for more than nine weeks before the surgical treatment of HER2-positive breast cancer and suggest a trastuzumab + pertuzumab + taxane or trastuzumab ± other chemotherapy regimen as a first-line treatment.
Despite the existence of these multiple guidelines and consensus regarding the initial treatment of HER2-positive advanced and recurrent breast cancers, no single standard treatment has been identified and currently available options mainly focus on “care” rather than “cure”. Therefore, to maximize therapeutic effectiveness, it is important to maintain the patient's quality of life (QOL) and ensure the long-term administration of therapy to the extent possible.
The ideal chemotherapeutic agent for use in a first-line trastuzumab-based combination therapy regimen for HER2-positive breast cancer remains under debate. One potential candidate is eribulin, a non-taxane-based inhibitor of microtubule dynamics derived from halichondrin B, a molecule with anti-tumor activity produced by the marine organism Halichondria okadai. Eribulin inhibits tubulin polymerization, thus suppressing microtubule functions and inducing cell-cycle arrest at the G2/M phase. It then induces apoptosis and shows tumor growth inhibitory action, and it is an agent that shows anticancer action by long-term inhibition of irreversible cell division (6-10). In vitro studies of eribulin demonstrated its growth-inhibitory effects in various human cancer cell lines, while in vivo studies demonstrated the remarkable anti-tumor effects of this molecule in various human cancer xenograft models.
In this clinical trial, the efficacy and safety of a combination regimen comprising eribulin and trastuzumab was evaluated as a first-line therapy for previously untreated progressive or recurrent HER2-positive breast cancer.
Patients and Methods
Study design. This multi-center, single-arm Phase II study was conducted to evaluate eribulin, an anticancer agent approved for patients with inoperable or recurrent breast cancers refractory to anthracycline and taxane chemotherapeutic drugs. On 11th November 2012, the institutional review board approved the study protocol, and all patients provided written informed consent. This study was registered with the UMIN Clinical Trial Registry (UMIN-CTR, UMIN000009890).
Eribulin dose reduction criteria and dose reduction level. (A) Eribulin dose reduction according to the worst value between the start of the previous cycle and the restart of administration. (B) If dose reduction beyond level 2 is needed, protocol treatment is discontinued.
Patients. The main eligibility criteria were used to select Japanese women aged ≥20 years who were histologically diagnosed with primary advanced or recurrent clinical stage IIIb, IIIc, or IV invasive breast cancer for which the HER2-positive status was confirmed by an immunohistochemistry (IHC) score of 3+ [or positive fluorescence in situ hybridization (FISH) result] in a biopsy test prior to registration. Patients who had previously been treated with anti-cancer drugs and trastuzumab as a primary therapy for progressive recurrent breast cancer, had active overlapping cancers with a disease-free period of <5 years, had brain metastases requiring treatment at the time of enrollment, or other serious diseases were excluded from the study. Cases deemed inappropriate for clinical trial registration by the attending physicians were also excluded.
Study treatment. Patients received eribulin at a dose of 1.4 mg/m2 intravenously (I.V.) on days 1 and 8 of each 21-day cycle. An initial trastuzumab dose of 8 mg/kg I.V. was administered on day 1, followed by a 6 mg/kg dose of trastuzumab on day 1 of each subsequent cycle. If eribulin could not be administered on day 8 because of an adverse event, the remaining number of days in the relevant cycle were recorded as a drug withdrawal and administration was resumed in the next cycle. Cycle initiation could be postponed until day 14, in which case eribulin and trastuzumab were administered on the same day as a rule. Postponement for more than 15 days led to discontinuation of the protocol treatment. The eribulin dose reduction criteria and dose reduction level were set according to Table I. In this study, it is thus notable that the termination of the protocol treatment was not a termination of the study. The image examination was to be performed every 3 months. The simultaneous outcome survey was decided two years after the final case registration.
Assessments
Efficacy. The primary endpoint was the RR (response rate), which was defined as the ratio of the number of subjects who began protocol treatment to the number of subjects for whom the best overall response was either a complete (CR) or partial (PR), according to the partially modified, Japan Clinical Oncology Group (JCOG) Japanese-language version of the revised Response Evaluation Criteria in Solid Tumors (RECIST), version 1.1. The tumor shrinkage effect was evaluated according to the JCOG Japanese-language translation of the New Evaluation Guideline for Response to Solid Cancer (RECIST guideline), revised version 1.1. The imaging modalities used to assess responses included computed tomography (CT) and magnetic resonance imaging (MRI). The secondary endpoints were defined as PFS (progression free survival), OS (overall survival), DOR (duration of response), CBRR (clinical benefit response rate), the frequency of adverse events.
Safety. Adverse events observed within 30 days after protocol treatment and protocol termination were evaluated based on the schedule determined by the treatment protocol. The maximum severity of each adverse event was entered in the case report for each cycle. Adverse events were evaluated using the Japanese-language version of the Common Terminology Criteria for Adverse Events (CTCAE), version 4.0. The events were scored as grade 1-4, depending on the definition that most closely met the observed event.
Sample size. The number of cases in this study was set based on the following two reports. In domestic Phase II study (11) using eribulin alone as the first-line, a response rate of about 35% was obtained as a result of the stratified analysis. Also, the response rate in global Phase II study (12) of trastuzumab combined chemotherapy in HER2 positive recurrent breast cancer is reported to be 59.3%. Based on these results, in this study, we considered that the threshold response rate of eribulin plus trastuzumab combination therapy was 35%, and the expected response rate was 60%. Also, assuming the significance level α=0.05 and the power 80% or more, the number of cases necessary for this test was calculated as 26 cases. Taking account of the 10% of analytical exclusion cases occurred during this study, the targeted number of cases in this study was 30 cases.
Statistical analysis. The patient group for the analysis of safety was a safety analysis set (SAS) that was a group excluding cases from the registered cases if no protocol treatment was performed at all. The group for the analysis of effectiveness was a full analysis set (FAS) that was a group excluding patients from SAS if they did not meet the inclusion criteria to this study or were unable to provide effectiveness data at all.
The primary endpoint was RR or the percentage of which we divided the number of cases where the best overall response was CR or PR in FAS by the number of cases in SAS. The 90% confidence interval (CI) was also calculated and whether the lower limit of the response rate was 35% or more was examined.
Secondary endpoints were PFS, DOR, OS, and CBRR. We estimated the PFS curve by the Kaplan–Meier method and calculated the median PFS time and the 95% CI. DOR and OS were analyzed in the same way as PFS. For CBRR, the percentage of cases in FAS where the best overall response was CR, PR, Long-SD (stable disease; ≥6 months) and the 90%CI were calculated, and we examined whether the CBRR was 35% or more. The incidence frequency and extent of adverse events were calculated by SAS, and the percentage of cases in which at least one adverse event was observed and their 95%CI. Also, the percentage of the cases in which adverse events of Grade 3 or higher were observed and the 95%CI were calculated, and the frequency of each grade was calculated for each type of adverse event.
SAS, version 9.4 (SAS Institute Inc., Cary, NC, USA) and R, version 3.1.0 (2014-04-10; R Project for Statistical Computing, Vienna, Austria) were used for analysis.
Results
Patient disposition. A total of were 28 patients were enrolled, and there were no cases of exclusion at the time of registration. The patient background is shown in Table II. The median age of patients was 62.5 years (range=40-80 years), the median height was 156.5 cm (range=145-167.9 cm), and the median weight was 54.5 kg (range=41.4-71 kg). Thirteen patients (46.4%) with inoperable advanced breast cancer and 15 patients (53.6%) with relapsed cases had surgery. All cases had HER2 overexpression, 17 out of them (60.7%) had ER-negative, 11 cases (39.3%) had ER-positive. ECOG PS (performance status) 0, 1, 2, 3 had 22 cases (78.6%), 3 cases (10.7%), 3 cases (10.7%), 0 cases, respectively.
Treatment was started for 28 patients, but 3 cases were excluded from FAS. The numbers of SAS and FAS were 28 and 25, respectively. One patient of the excluded three patients was transferred to another hospital, so data on the effectiveness after starting protocol treatment could not be obtained. The remaining two patients had brain metastasis requiring treatment and violated the exclusion criteria. One of the two patients with brain metastasis was not able to terminate the protocol treatment at the time of the simultaneous outcome survey. We evaluated the patient data for the safety of the protocol treatment only.
Ten cases had exacerbations of the tumor which was the most common reason for termination of the protocol treatment following post-treatments. Subsequently, there were 5 cases of termination of treatment appealed from patients (due not to adverse events), 4 cases of termination due to adverse event and doctor's decision, respectively, 3 cases of termination of treatment appealed from patients (due to adverse events), one case of a transfer to another hospital. We continued observations for these patients during the post-treatment until progression or death appeared.
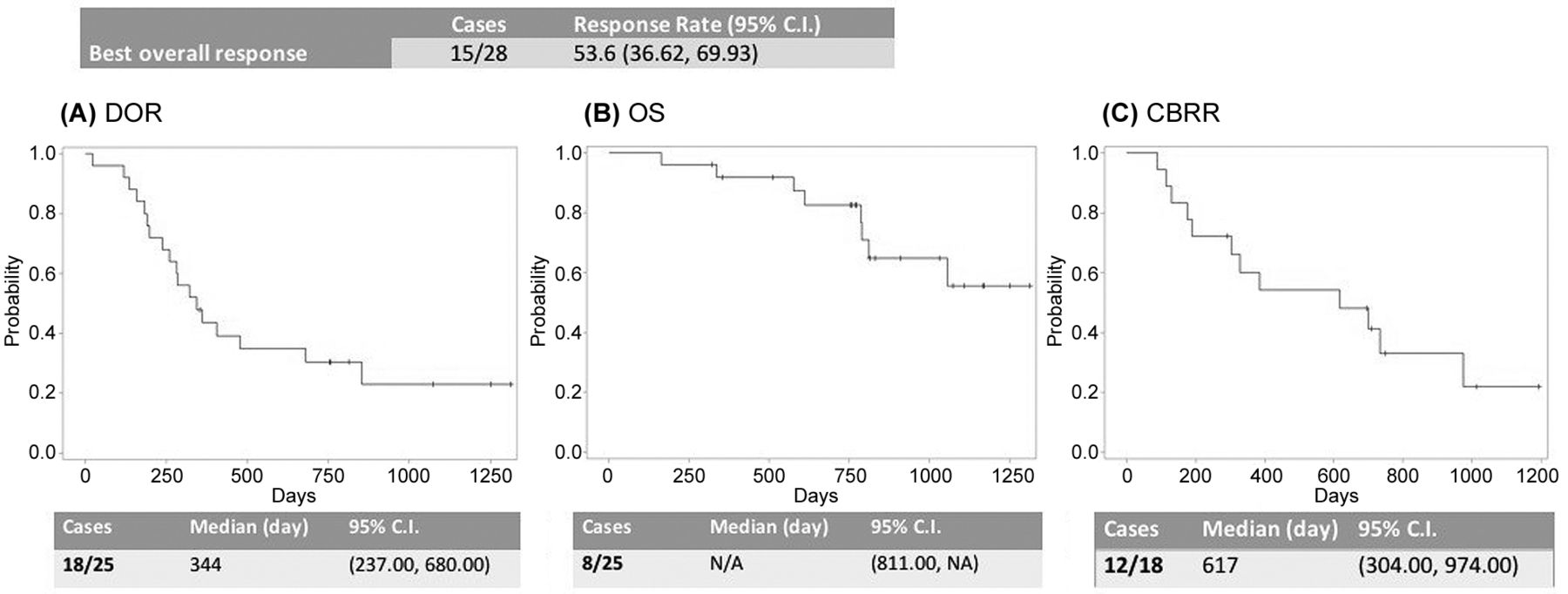
Efficacy. The primary endpoint, best overall response, was 53.6% in 15 cases out of 28 (CR: 4 cases, PR: 11 cases in FAS). RR was 53.6% when the denominator was taken as the number of SAS, and the lower and upper limits of the 90%CI were 36.6% and 69.9%, respectively. As a result, the lower limit of the response rate exceeded the initially set threshold value of 35%. The median of the secondary endpoints, DOR, was 344 days and the lower and upper limits of the 95% CI were 237 days and 680 days, respectively (Figure 1A). As 17 out of 25 cases survived at the time of simultaneous outcome, OS did not fall below the survival probability of 0.5 (Figure 1B). DOR was targeted at 18 subjects whose total effect was judged to be CR or PR by the attending physician. The event was accepted in 12 cases; the median response duration was 617 days, the lower and upper limits of the 95%CI were 304 days and 974 days (Figure 1C), respectively.

Primary endpoint (Overall best response) and secondary endpoints (DOR, OS and CBRR).
According to the Central Judgment Committee, the CBRR included 4, 11, and 1 case that achieved a CR, PR, and Long-SD, respectively, for a total of 16 cases. Therefore, the CBRR was 64.0%, with lower and upper 90%CI limits of 45.61% and 79.76%, respectively.
Additional analyses. In Figure 2, the spaghetti plots depict the cycle number for each case on the horizontal axis and the PS on the vertical axis. Here, the PS tended to improve as the number of treatment cycles increased. Figure 3 depicts graphs in which the number of treatment cycles for each case are plotted on the abscissa while the total effect judgment [CR, PR, SD, PD (progressive disease), NE (not evaluable)] are plotted on the vertical axis. This graph indicates that CR and PR were achieved at an early stage after protocol treatment initiation.
Safety
Exposure. An average of 14 cycles of protocol treatment were administered (range=9.8-53 cycles). Throughout the dosing cycle, eribulin was administered on days 1 and 8 (i.e., according to the protocol) in 94.3% of cycles. Day 1 eribulin administration was postponed in 11.7% of cycles. The eribulin dose was decreased from the previous cycle in 3.4% of cycles, and eribulin administration on day 8 was skipped in 5.7% of cycles. No events of eribulin dose reduction on day 8 were recorded. Trastuzumab was administered in all cycles.
Twenty-three cases (92.0%) received post-treatment, and the most frequent post-treatment was chemotherapy (20 cases, 80.0%). In addition, 7 patients underwent surgery for stage I (3 cases), IIA (2 cases), IIIC (1 case), and IV tumors (1 case). The most common post-treatment was trastuzumab (16 cases, 64.0%). The molecular targeted drugs bevacizumab and lapatinib were also administered as post-treatment to 2 patients (8%). Protocol treatment was stopped because of exacerbation in 3 of 4 patients receiving molecular targeted drugs, while the fourth case was discontinued because of adverse events.
Adverse events. Table III summarizes the grade levels of all adverse events, which were observed in 26 of 28 cases (92.9%). Grade ≥3 adverse events were observed in 12 cases (42.9%). The most frequent adverse event was peripheral sensory neuropathy, which was observed in 19 cases (67.9%). Table IV presents the clinical laboratory values corresponding to the grade levels of adverse events. The most frequent adverse event was white blood cell count reduction in 14 of 28 patients (50.0%). No febrile neutropenia or cardiac dysfunction events were observed.
Discussion
In the analysis of PFS, cases where no event was observed, and cases of withdrawing or dropping protocol treatment due to reasons other than exacerbation were discontinued on the survey date of the simultaneous outcome survey. However, in the cases that were not traceable in the simultaneous outcome survey, the evaluation day of the last tumor evaluation performed was set as the censored date. Also, 1 case was unable to follow up with a simultaneous outcome study. Data collection was performed well, there were no data deficiencies, except for cases of non-traceable cases, regarding patient background, treatment at registration, treatment situation, adverse event investigation. The intermediate analysis was not specified in the protocol, and it was not carried out. TRI (Translational Research Informatics Center, Kobe, Japan) data center appropriately inquired and confirmed the doubt of the data entered during the research period.

Trends of ECOG PS. Improvement of PS was observed in many cases with the course of treatment.
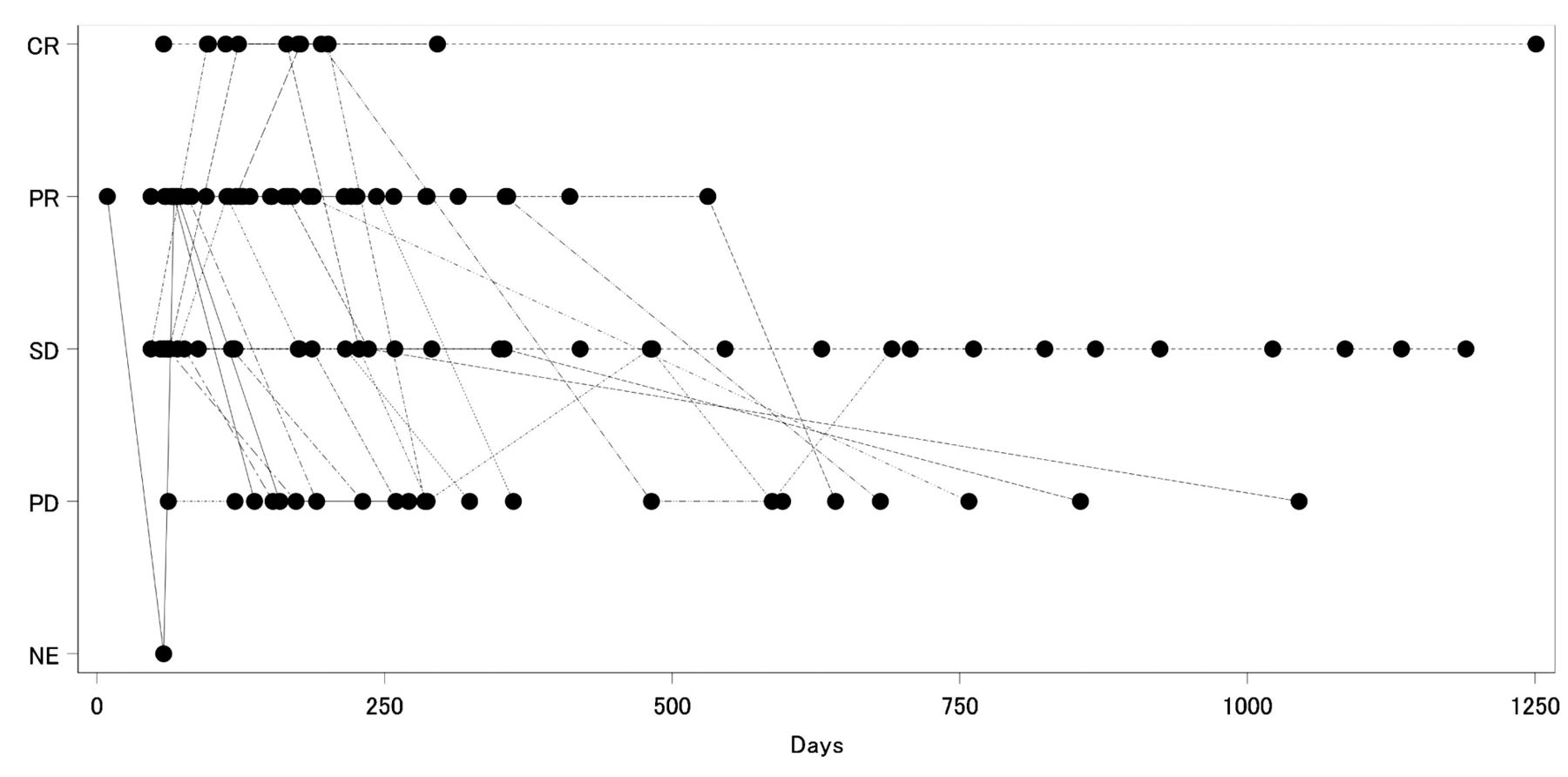
Overall best response. The day on which CR or PR was first determined, all cases were before 314 days after the start of protocol treatment.
After termination of protocol treatment, ten patients received molecular targeted drugs (bevacizumab, lapatinib, pertuzumab) (Table V). We discussed whether this administration affected the evaluation in the PFS and OS of the secondary endpoint. Five out of ten patients discontinued the protocol treatment due to progression and received the post-treatment; they, therefore, did not affect the evaluation of PFS. Two of ten patients, however, received the post-treatment due to adverse events during the protocol treatment period and died after 127 days and 519 days after the protocol treatment was terminated. We were unable to deny the possibility that these two patients affected the PFS. Also, at the time of the simultaneous outcome survey, six out of ten cases had deceased, but four cases were alive. Accordingly, the administration of other molecular targeted drugs as post-treatment could also have affected OS.
Baseline demographics and disease characteristics of enrolled Japanese patients with HER2+BC.
Regarding outcomes, a RR (primary endpoint) of 53.6% was observed, with a lower limit that exceeded the initially set threshold of 35%. Therefore, we deemed this combination regimen of eribulin + trastuzumab to be “efficacious”.
In the Eisai Metastatic Breast Cancer Study Assessing Physician's Choice Versus E 7389 (EMBRACE) trial (13) of 762 patients with locally recurrent or metastatic breast cancer, the researchers compared OS between an eribulin-treated group and a primary care physician-selected treatment group and found that the former had an OS prolongation of 2.7 months (13.2 vs. 10.5 months, HR=0.81, p=0.014). In addition, this trial reported RRs (CR + PR) of 4.7% in the primary care physician-selected treatment group and 12.2% in the eribulin administration group, thus demonstrating that the effect of eribulin exceeded those of other drugs. Furthermore, a Japanese Phase II trial of eribulin monotherapy (11), in which 80 Japanese patients pretreated with an anthracycline and a taxane, yielded a RR (CR + PR) of 21.3%, SD rate of 37.5%, clinical benefit rate (CR + PR + SD>6 months) of 27.5%, and RR of 36% among patients who received 0 or 1 pretreatment regimens. Although differences in patient backgrounds prevent a simple comparison of our research with these previous studies, the RR in our study was not inferior and we observed no tendency toward an increase in cardiac events with the addition of trastuzumab to eribulin.
Total adverse events in eribulin plus trastuzumab combination therapy protocol treatment. There was no evidence of febrile neutropenia nor cardiac dysfunction.
Laboratory values: The most frequent adverse event was white blood cell count reduction. Adverse events of Grade 3 or higher were observed in 20% of leukopenia and neutropenia.
In a Phase II study of a combination regimen of eribulin and trastuzumab in 52 patients with metastatic breast cancer who previously received ≥2 chemotherapy regimens in the metastatic setting (14), Puhalla et al. reported a RR of 59.3% and PFS of 9.2 months, as well as no increase in synergistic adverse events due to the combined regimen. Although that study enrolled patients who had previously received trastuzumab, the efficacy and safety results were comparable to those of our study. By contrast, our RR exceeded that of a Japanese study reported by Araki et al. (15), which yielded a RR of 34.8%. Our study also reported a superior median PFS of 49.1 weeks (344 days, 11.5 months), compared to the 42.6 weeks reported by Araki et al. However, Araki and colleagues did not define clinical stages, and therefore was not possible to directly compare the patient populations of the two studies. Furthermore, Araki et al. enrolled patients who had been previously treated with taxane and trastuzumab. Accordingly, the increased clinical efficacy observed in our study may be attributable to the focus on first-line treatment for relapse.
Patients received molecular targeted drugs after termination of protocol treatment.
Although Araki et al. did not discuss OS, Wilk et al. (12) evaluated the efficacy and safety of eribulin plus trastuzumab as a primary treatment for HER2-positive recurrent and metastatic breast cancer and reported a RR of 71.2% and median PFS of 11.6 months. Although our study reported a higher RR, the median PFS durations of the two studies were very similar. The CLEOPATRA trial (16) reported a RR of 80.2% and median DOR of 20.2 months (range=16-24 months) among treatment-naïve patients who received a combination of trastuzumab + pertuzumab + docetaxel for HER2-positive progressive recurrent breast cancer. Although these outcomes were superior to those in our study, the authors reported the incidence of febrile neutropenia in 13.8% of cases and cardiac disorder in 9.6% of cases. By contrast, our study observed no cases of either adverse event. Rather, we most frequently observed reductions in the white blood cell count, similar to related previous studies (12, 14, 15, 17). Furthermore, in post hoc analysis of CLEOPATRA study, Miles et al. (18) evaluated the relationship between docetaxel (D) treatment cycles and clinical outcomes after accounting for HER-2 targeted therapy. As a result, in fewer than 6D cycles group, 100% of patients discontinued D and 25.9% of patients discontinued all study treatment because of adverse events. Also, fewer than 6D cycles was associated with poorer PFS and OS compared with 6D cycles.
Conclusion
No major problems were observed with the tolerability of eribulin plus trastuzumab during the study period, as indicated by the few deviations and changes in protocol treatment. Taken together with the safety data, this suggests that a combination regimen of eribulin plus trastuzumab may be safer for elderly patients and those with systemic diseases or cardiac dysfunction and would, thus, be economically advantageous.
In conclusion, our results indicate that a combination regimen of eribulin plus trastuzumab is a potentially important primary treatment option for advanced and recurrent HER2-positive breast cancer, with good safety and efficacy outcomes.
Acknowledgements
The Authors would like to thank Kenichi Kono and Akina Taji (TRI, Kobe) for performing data collection and Hiromi Tsukada (TRI, Kobe) for performing the data analysis. The Authors also thank Fumine Tsukamoto (JCHO Osaka Hospital, Osaka), Seung J. Kim (Osaka University, Osaka), and Wataru Sakamoto (Osaka University, Osaka) from the independent data monitoring committee, and Yasuhiro Tamaki (International Cancer Center, Osaka), Katsuhide Yoshitomi (Osaka Police Hospital, Osaka), and Yoshifumi Komoike (Kinki University, Osaka) from the central judgment committee. We would like to thank Editage (www.editage.jp) for English language editing.
Footnotes
Conflicts of Interest
The Authors declare no conflicts of interest relevant to this article.
- Received May 23, 2018.
- Revision received June 10, 2018.
- Accepted June 12, 2018.
- Copyright© 2018, International Institute of Anticancer Research (Dr. George J. Delinasios), All rights reserved
In this issue





{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.