


Abstract
Background: Pancreatic cancer is a highly lethal malignancy with a poor prognosis. This study was set up to investigate the combined effect of gemcitabine and fisetin, a natural flavonoid from plants, on human pancreatic cancer cells. Meterials and Methods: Cytotoxic effect of fisetin in combination with gemcitabine on MiaPaca-2 cells was evaluated by the MTT assay, caspase 3/7 assay and propidium iodide/Annexin V. Extracellular signal-regulated kinase (ERK)-v-myc avian myelocytomatosis viral oncogene homolog (MYC) pathway was investigated by western blotting and quantitative real-time polymerase chain reaction. Results: Combination treatment with fisetin and gemcitabine inhibited the proliferation of pancreatic cancer cells within 72 h and induced apoptosis, as indicated by activation of caspase 3/7. Fisetin down-regulated ERK at the protein and mRNA levels, and reduced ERK-induced MYC instability at the protein level. Conclusion: Fisetin sensitized human pancreatic cancer cells to gemcitabine-induced cytotoxicity through inhibition of ERK-MYC signaling. These results suggest that the combination of fisetin and gemcitabine could be developed as a novel potent therapeutic.
Pancreatic cancer is the fourth leading cause of cancer-related death in the Western world. It has an extremely poor prognosis, with a 5-year relative survival rate of 5% and a median survival of 3.5 months for patients with non-resectable tumors (1). Surgical resection is the only potentially curative treatment, although relapses are common even in these cases. Gemcitabine, a nucleoside analog of cytidine, is the standard systemic treatment for pancreatic cancer. Nonetheless, the efficacy of gemcitabine as a single agent remains modest, with a median survival of approximately 6 months in randomized trials and a 12-month survival rate of less than 20% in gemcitabine-treated patients (2). Two combination treatments, gemcitabine in combination with nab-paclitaxel or erlotinib, have a clinical benefit, although the median survival still remains less than 12 months (3, 4). Therefore, the development of efficient therapeutic strategies is critical to improve the clinical management and prognosis of patients with pancreatic cancer.
Fisetin [2-(3,4-dihydroxyphenyl)-3,7-dihydroxy-4H-1-benzopyran-4-one] is a naturally-occurring flavonoid and antioxidant present in various fruits and vegetables. Fisetin induces cell-cycle arrest with/without caspase-dependent apoptosis in triple-negative breast cancer (5), colon cancer (6), and non-small lung cancer (7). Fisetin exerts an inhibitory effect on adhesion, migration, and invasion in A549 human lung cancer cells by inhibiting the phosphorylation of extracellular signal-regulated kinases 1 and 2 (ERK1/2) and down-regulating the expression of matrix metalloproteinase-2 and urokinase plasminogen activator at the protein and mRNA levels (8). In addition, fisetin treatment negatively regulates the growth of chemoresistant AsPC-1 pancreatic cancer cells by suppressing death receptor 3 (DR3)-mediated nuclear factor (NF)-ĸB activation (9).
Rat sarcoma (RAS) is a small GTP-binding protein and the common upstream molecule of several signaling pathways including rapidly accelerated fibrosarcoma (RAF)/mitogen-activated protein kinase kinase (MEK)/ERK (10). ERKs, which act as downstream effectors of RAS signaling, are serine/threonine kinases, the activity of which is positively regulated by phosphorylation mediated by MEK1/2. MEKs can phosphorylate ERK1 on T202 and Y294, and ERK2 on T185 and Y187, to activate their kinase function (11). ERKs can directly phosphorylate a set of transcription factors including activating protein 1 (AP1), E26 transformation-specific sequence-1 (ETS1), NF-ĸB, and v-Myc myelocytomatosis viral oncogene homolog (c-MYC), which were initially identified as proto-oncogenes (12). Moderate c-MYC expression induces cell proliferation by promoting an increase in cell mass and by modulating the expression of genes regulating cell cycle progression (13). Overexpression of c-MYC by aberrant activation of this pathway induces tumorigenesis (13). The stability of the c-MYC protein is markedly increased by activated ERKs as a result of direct phosphorylation of Ser 62 on MYC (14). The proto-oncogene c-MYC is a master regulator of cancer cell survival in many pancreatic cancer cell lines including MiaPaca-2 (15). Pancreatic cancer displays the highest frequency (70-95%) of somatic activating mutations in the RAS gene (16). A homozygous mutation on codon 12 of the RAS gene in Capan 1 and MiaPaca-2 pancreatic cancer cells leads to high levels of MEK and ERK phosphorylation (17). The modulation of c-MYC expression through the RAS-ERK signaling pathway in cancer cells might be a promising target for cancer therapy.
Herein, we explored the mechanism underlying the anticancer effects of fisetin on MiaPaCa-2 human pancreatic cancer cells through the RAS-ERK signaling pathway and the efficacy of combination therapy with fisetin and the conventional pancreatic cancer drug gemcitabine.
Materials and Methods
Cells and reagents. The MiaPaca-2 human pancreatic carcinoma cell line was obtained from the Korean Cell Line Bank (KCLB, Seoul, Korea). MiaPaca-2 cells were grown in Dulbecco's modified Eagle's medium (DMEM, Welgene, Gyeongsan, Korea) with 1% streptomycin and penicillin (Corning, Corning, NY, USA), and 10% fetal bovine serum (Tissue Culture Biologicals, Tulare, CA, USA) at 37°C and 5% CO2 in a humidified environment. Compounds [50 μM fisetin (TOCRIS, Bristol, UK) with/without 100 nM gemcitabine (Yuhan, Seoul, Korea)] were added, and the cells were incubated as described below. Cells were treated with dimethyl sulfoxide (DMSO) (0.25%) as a control.
Polyclonal antibodies against c-MYC, phospho-ERK1/2 and ERK1/2 were purchased from Cell Signaling Technology, Inc. (Beverly, MA, USA). Polyclonal α-tubulin (LF-PA0146) was purchased from Abfrontier (Seoul, Korea). Horseradish peroxidase (HRP)-conjugated donkey anti-rabbit IgG Ab (sc-2004) was purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA).
3(4,5-Dimethylthiazol-2yl)-2,5diphenyltetrazoliumbromide (MTT) assay. Cells were seeded on 96-well plates in a final volume of 100 μl/well at a density of 4×103 cells/well, incubated for 24 h at 37°C, and treated with fisetin with/without gemcitabine for 24, 48, and 72 h. To determine cell viability, 10 μl of 5 mg/ml MTT (Promega, Madison, WI, USA) in phosphate-buffered saline (PBS) was added to the cells and allowed to develop for 1 h at 37°C. To dissolve formazan crystals, 100 μl of 10% sodium dodecyl sulfate (SDS) solution was added to each well and the plates were incubated overnight at room temperature. The solution was mixed to ensure complete solubilization. Colorimetric measurements were taken at 570 nm by Sunrise™ (Tecan, Mannedorf, Switzerland).
Apoptosis detection. Apoptosis induction was examined by assessing caspase activity. Cells were seeded on 96-well plates containing a final volume of 100 μl/well at a density of 7×103 cells/well, incubated for 24 h at 37°C, and treated with fisetin with/without gemcitabine for further 24 h. Caspase 3/7 activity was quantified using the Caspase-GloR 3/7 assay according to the manufacturer's instructions (Promega) and measured on a luminometer (VICTOR X; Perkin Elmer, Waltham, MA, USA). Background luminescence (cell culture medium without cells and Caspase-Glo 3/7 reagent) was subtracted from all measurements. Luminescence values of non-treated cells (negative control) were calculated as fold increases in caspase activity of samples. Experiments were performed in triplicate.
Apoptosis was examined using an allophycocyanin-labeled Annexin V/Propidium Iodide (PI) Apoptosis Detection kit (BD Biosciences, San Jose, CA, USA). Cells (1×105) were suspended in 100 μl Annexin-binding buffer (10 mM HEPES/NaOH, pH 7.4, 140 mM NaCl, and 2.5 mM CaCl2). The cell suspension was stained with 5 μl allophycocyanin-conjugated annexin V and 5 μl PI for 15 min at room temperature, and then 400 μl annexin-binding buffer were added. Apoptotic and necrotic cells were analyzed with a flow cytometer (FACS Canto II; BD Biosciences). Results were analyzed with FlowJo software (Ashland, OR, USA).
Western blot analysis. Cells were harvested after exposure to fisetin with/without gemcitabine for 24 h, washed twice in PBS, and then suspended in 20 μl lysis buffer (50 mM Tris-HCl, pH 8.0, 5 mM EDTA, 150 mM NaCl, 1% NP-40, 1 mM phenylmethylsulfonyl fluoride, 0.1% SDS, protease inhibitor cocktail, 5 mM NaF, and 2 mM Na3VO4). Cell suspensions were kept on ice for 15 min and then centrifuged at 12,000 rpm for 15 min at 4°C. Protein concentrations were determined using BCA Protein Assay kit (Thermo Fisher Scientific Korea, Seoul, Korea) following the manufacturer's instructions. Equal amounts (30 μg) of protein were loaded into each lane, separated by SDS-polyacrylamide gel electrophoresis (PAGE) on a 10% polyacrylamide gradient gel, and then transferred onto polyvinylidene difluoride membranes (Amersham Biosciences, Piscataway, NJ, USA). Membranes were blocked with 3% bovine serum albumin in PBS containing 1% Tween-20 (PBS-T) for 30 min at RT, followed by incubation with primary antibodies overnight at 4°C. Membranes were washed three times in PBS-T and incubated for 1 h at room temperature with HRP-conjugated secondary antibodies. Bands were visualized using an enhanced chemiluminescence detection system (ImageQuant LAS 4000; GE Healthcare, Uppsala, Sweden). Quantification of relative band intensity was performed with Image Studio™ Lite Ver. 5.2 (Li-COR Bioscience, Lincoln, NE, USA).
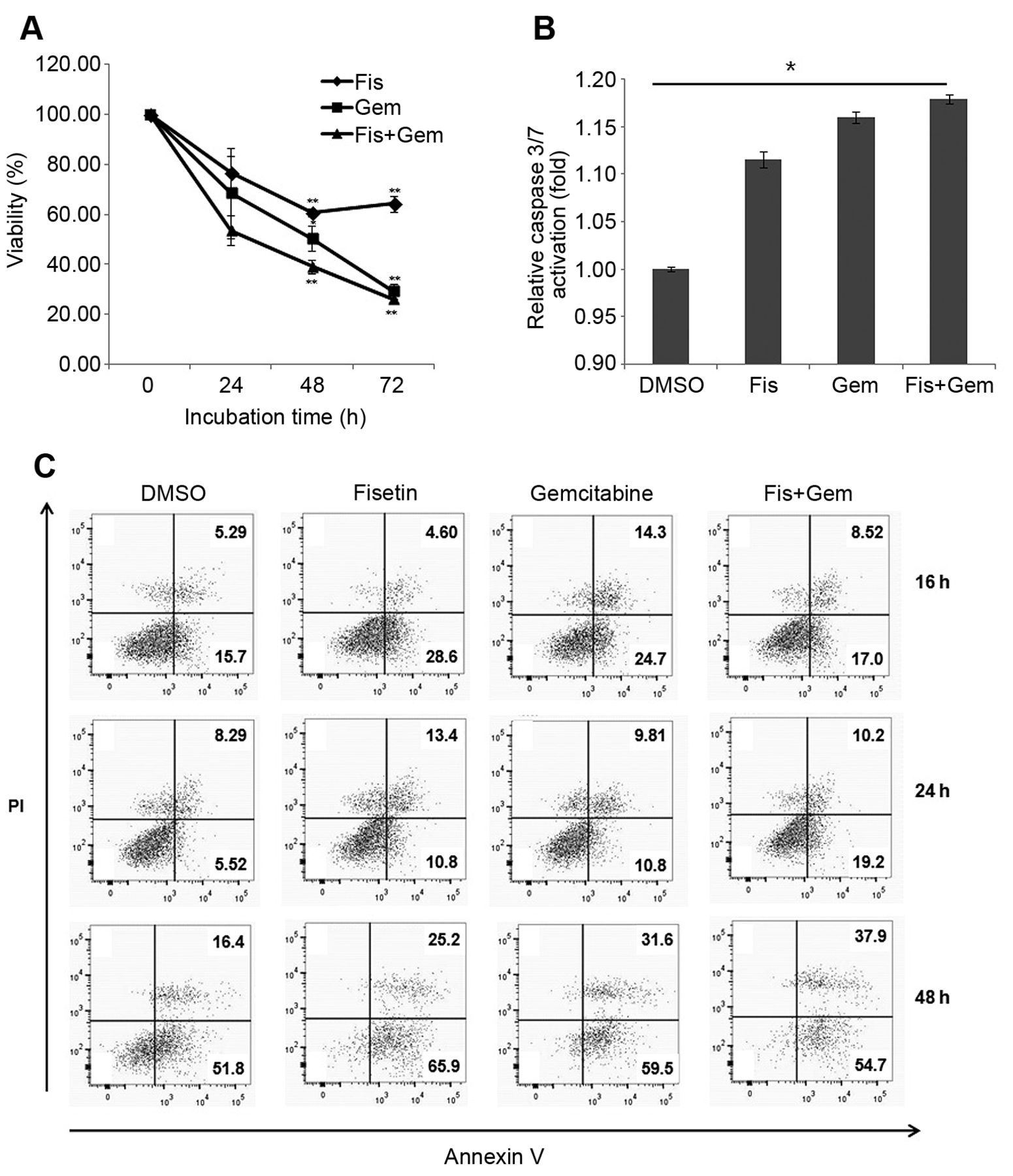
Fisetin (Fis) increases the cytotoxicity of gemcitabine (Gem). A: The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay showed that fisetin and gemcitabine reduced numbers of MiaPaca-2 cells in a time-dependent manner. The viability was calculated as the percentage viability relative to those of vehicle control (dimethyl sulfoxide;DMSO)-treated cells. The results are representative of three independent experiments performed in triplicate (mean±SEM). Significantly different at *p<0.05 and **p<0.01, compared with DMSO treatment. B: Fisetin and gemcitabine induced caspase 3/7-dependent apoptosis at 24 h. C: MiaPaca-2 cells were stained with annexin V and propidium iodide (PI) and analyzed by flow cytometry after treatment with fisetin with/without gemcitabine for the indicated times. The results are representative of three independent experiments.
Quantification of mRNA levels by real-time polymerase chain reaction (RT-qPCR). Total RNA was extracted using the TRIzol™ (Thermo Fisher Scientific, Waltham, MA, USA) reagent following the manufacturer's instructions. The quantity and purity of total RNA were measured using a UV/Vis-Spectrophotometer (NanoDrop ND-2000; Thermo Fisher Scientific). The cDNA was synthesized from 0.7 μg RNA using SuperScript III RT (Invitrogen; Thermo Fisher Scientific, Waltham, MA, USA). The amplification was performed in 20 μl containing 1 μl of each primer (5 pmol), SYBR Green PCR master mix (Applied Biosystems, Foster City, CA, USA), and 2 μl of 1:10 diluted cDNA. Amplification conditions were 95°C for 10 s and 60°C for 1 min for 40 cycles. Primer sequences were as follows: MYC: forward: 5’-AAA GGC CCC CAA GGT AGT TA-3’, and reverse: 5’-TTT CCG CAA CAA GTC CTC TT-3’ (18); ERK1: forward: 5’-CAGGAGACAGCACGCTTCCAG-3’, reverse: 5’-TCTAACAGTCTGGCGGGAGAGG-3’; ERK2 forward: 5’-CAACCCACACAAGAGGATTGAA-3’, reverse: 5’-GTCGAACTTGAATGGTGCTTCG-3’ (19); glyceraldehyde-3-phospahte dehydrogenase (GAPDH) forward: 5’-ACC CAG AAG ACT GTG GAT GG-3’, reverse: 5’-TTC AGC TCA GGG ATG ACC TT-3’. Primers for MYC and GAPDH were synthesized by Bioneer (Daejeon, Korea). Primers for ERK1 and ERK2 were synthesized by Macrogen (Seoul, Korea). Assays were run on an Applied Biosystems 7500 Real-Time PCR system, and results were analyzed by the standard curve method with RQ manager 1.2.1 software (Applied Biosystems, Lincoln, CA, USA).
Statistical analysis. Student t-test was performed using Excel software (Microsoft, Seattle, CA. USA). For caspase assay, Kruskal–Wallis test (nonparametric ANOVA) with Dunn's multiple comparisons test was performed using a GraphPad InStat ver. 3 software (GraphPad Software, Inc., San Diego, CA, USA).
Results
Fisetin increased the cytotoxicity of gemcitabine by inducing caspase-dependent apoptosis. The effect of combination treatment with fisetin and gemcitabine on the growth of MiaPaca-2 cells in vitro was assessed. Each agent was used at a concentration that induced moderate cell death (data not shown) to determine the combinatory effect. Cells were treated with fisetin and gemcitabine separately or in combination, and growth rates were assessed using the MTT assay after 72 h of treatment. Representative data are shown in Figure 1A. Combination treatment significantly inhibited growth compared with vehicle control (DMSO) cells at 48 h (p<0.05, Figure 1A). The cytotoxicity of combination treatment was 60.9% at 48 h, compared with 49.6% for gemcitabine alone. However, at 72 h, the effect of combination treatment was comparable to that of gemcitabine alone. These observations suggest that fisetin sensitized cells to gemcitabine within 2 days.
To investigate whether the suppression of proliferation shown in Figure 1 was mediated by the induction of apoptosis, caspase 3/7 activity was measured. In MiaPaca-2 cells, caspase 3/7 activity increased in response to single-agent treatment compared with that in controls, whereas combination treatment with fisetin and gemcitabine increased caspase 3/7 activity by 1.15-fold over that in the controls (Figure 1B). The increase was statistically significant. To further explore the effect of gemcitabine and fisetin on cellular apoptosis, the apoptosis rate was determined by annexin V/PI staining. The data indicate that treatment with fisetin and/or gemcitabine had a time-dependent effect on apoptotic cell death. The rates of apoptosis in MiaPaca-2 cells treated with DMSO, fisetin, gemcitabine, and combination treatment were 5.52%, 10.8%. 10.7%, and 19.2%, respectively, at 24 h. Although there was no significant apoptotic induction at 16 or 24 h of treatment, combination treatment induced apoptosis more effectively at 24 h than single-agent treatments. The results at 24 h of treatment were consistent with those obtained in the caspase 3/7 activity assay. After 48 h of treatment, fisetin and gemcitabine induced a higher rate of MiaPaca-2 cell apoptosis and cytotoxic effects at both early (annexin V+PI−) and late (annexin V+PI+) stages according to the annexin V/PI staining analysis (Figure 1C). The cytotoxicity of combination treatment was 92.6% (early apoptosis=54.7%, late apoptosis=37.9%).

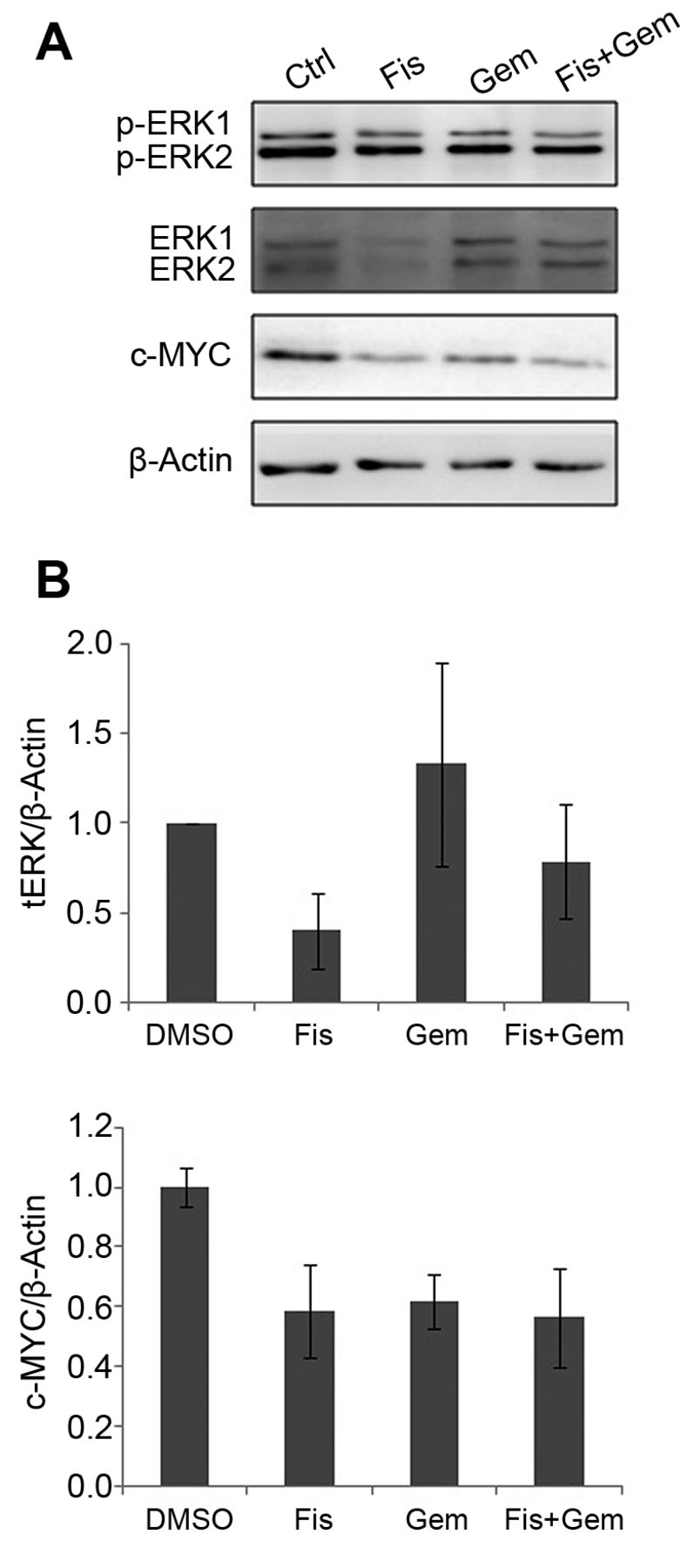
Fisetin (Fis) down-regulates extracellular signal-regulated kinases 1 and 2 (ERK1/2) and v-Myc myelocytomatosis viral oncogene homolog (c-MYC) at the protein level. A: Cells were treated with fisetin with/without gemcitabine (Gem) as described in Materials and Methods, and cell signaling-associated proteins were measured by western blot analysis. The protein levels of phospho-(p)-ERK, total (t) ERK, c-MYC, and α-tubulin were examined using sodium dodecyl sulfate-polyacrylamide gel electrophoresis and western blot analysis. The results are representative of three independent experiments. B: Quantification of band intensities shown in (A). The experiments were performed in triplicate. Data represent the mean±SEM of three independent experiments.

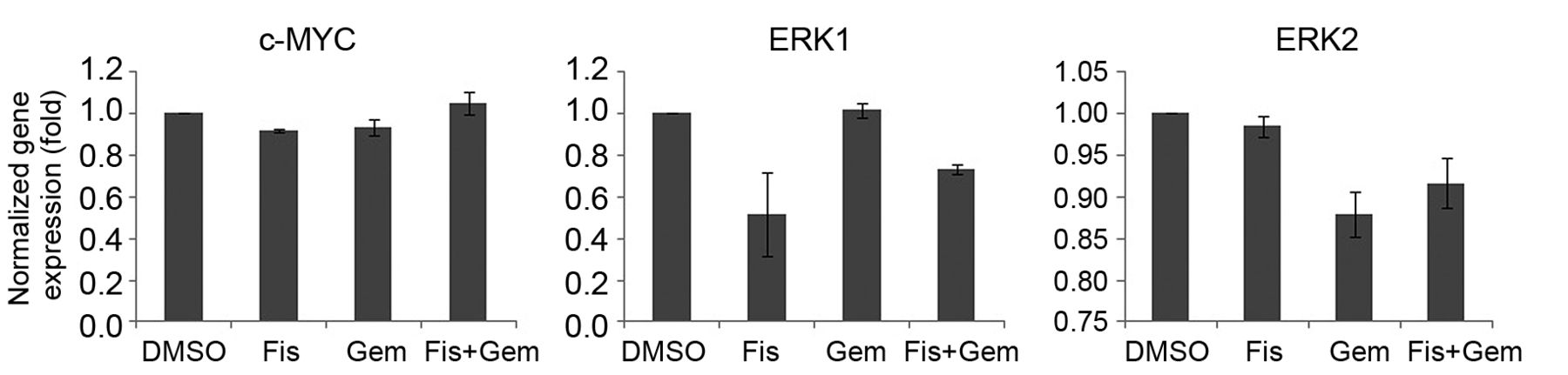
Fisetin (Fis) down-regulates extracellular signal-regulated kinases 1 (ERK1), but not v-Myc myelocytomatosis viral oncogene homolog (c-MYC), at the transcriptional level. Reverse transcription-quantitative polymerase chain reaction results from MiaPaCa-2 cells treated with fisetin with/without gemcitabine (Gem) showing MYC, ERK1, and ERK2 mRNA levels. The mRNA levels of c-MYC, ERK1, and ERK2 were normalized to glyceraldehyde-3-phospahte dehydrogenase (GAPDH). Each experiment was repeated in triplicate, and the results are presented as the mean±SEM.
Fisetin prevented ERK-mediated stabilization of newly-translated MYC protein. To investigate the mechanism by which fisetin enhanced the inhibition of cell growth of MiaPaca-2 cells, the signaling pathways involved in pancreatic cancer were explored. Fisetin treatment reduced total ERK1/2 levels to 40×21.1% of the control, whereas phosphorylated ERK1/2 levels remained constant (Figure 2A and B). To determine whether the down-regulation of ERKs was linked to c-MYC protein stability, the protein level of c-MYC was analyzed. As shown in Figure 2A and C, fisetin reduced c-MYC protein levels to 66.6×15.5% of the vehicle control. These observations suggest that the activation of newly synthesized ERK1/2 is involved in c-MYC stability but not the constitutive phosphorylation of ERKs.
Fisetin down-regulated ERK1 without affecting c-MYC mRNA level. To understand the mechanisms underlying the decrease in ERKs and c-MYC expression by fisetin, RT-qPCR was performed. Fisetin reduced the mRNA level of ERK1 to 51.7×20.3% of the DMSO control, whereas it had no significant effect on the mRNA level of c-MYC (92.1×0.7%) and ERK2 (98.4×1.2%) in MiaPaca-2 cells (Figure 3). The results suggest that down-regulation of ERK1 at the transcriptional level caused the instability of c-MYC protein (Figure 4).
Discussion
In this study, we showed that combination treatment with fisetin and gemcitabine prevented the proliferation of MiaPaca-2 pancreatic cancer cells, which express a mutated version of RAS (17). Fisetin in combination with gemcitabine also induced apoptosis in a caspase-3/7-dependent manner. The results support a recent report where fisetin induced caspase-3, -8, and -9 (20). Fisetin down-regulated ERK expression in MiaPaca-2 cells at the protein and mRNA levels. The levels of phosphorylated MEK and ERK in the MiaPaca-2 cell line are high, presumably because of a homozygous mutation on codon 12 of the K-RAS gene (17). The expression of c-MYC decreased in this pancreatic cancer cell line at the protein level on treatment with fisetin. These data are in line with the notion that c-MYC is a downstream target of the RAS–ERK pathway and suggest that aberrant growth of pancreatic cancer cells can be halted by targeting c-MYC after ERK down-regulation by fisetin. We found that the c-MYC levels were correlated with ERK expression levels in pancreatic cancer cells. The down-regulation of ERK1 at the transcriptional level induced c-MYC instability at the protein level, presumably as a result of the failure of phosphorylation of Ser 62 on c-MYC.

A proposed mechanism of the anticancer effect of fisetin on pancreatic cancer cells. Reduction of extracellular signal-regulated kinases 1 (ERK1) mRNA level by fisetin leads to v-Myc myelocytomatosis viral oncogene homolog (c-MYC) protein reduction and pancreatic cancer cell death.
Several genetic mutations occur with high frequency in pancreatic cancer, including K-RAS mutation; loss of p16, p53 and deleted in pancreatic carcinoma, locus 4 (DPC4) function; and overexpression of multiple receptor tyrosine kinases (21). These alterations up-regulate RAS–ERK activity, a process that stimulates proliferation and enhances survival-related responses including drug resistance (22). The RAS–ERK pathway is considered an attractive target for the development of pancreatic cancer therapies. The expression levels of mutant K-RAS and c-MYC are equally important for the initiation and maintenance of pancreatic cancer (23). RAS-mediated control of c-MYC protein occurs through the action of the RAF–ERK signaling pathway in promoting the stabilization of MYC by phosphorylation of Ser 62 in the N-terminal domain of MYC (14, 24). Conversely, the decrease in the MYC protein is caused by glycogen synthase kinase-3 (GSK-3)-mediated phosphorylation at Thr 58 through the phosphatidylinositol 3-kinase (PI3K)/protein kinase B (AKT) signaling pathway (14). RAS activates AKT, which in turn inactivates GSK-3 and blocks the c-MYC degradation pathway (14). In addition, fisetin slightly reduced the AKT protein level (data not shown), suggesting that activated GSK-3 may also lead to c-MYC degradation.
High ERK protein expression levels are correlated with shorter survival in patients with triple-negative breast cancer (25), while activation of ERK induced apoptosis in human gastric and breast cancer cells by the natural products equol and wogonin (26, 27). A previous study showed that c-MYC expression and phosphorylation are reduced in ERK1/ERK2 or ERK2 siRNA-transfected human muscle-derived rhabdomyosarcoma cell lines to a level similar to that in MEK/ERK inhibitor-treated cell lines (28). Reduced expression of RAS-ERK–signaling pathway components could be an important therapeutic target in cancer cells. Hence, the effect of fisetin on down-regulating ERK and c-MYC may help eliminate pancreatic cancer cells. However, recent reports indicate that the anticancer effect of fisetin may be mediated by different pathways in a cell type-dependent manner. In laryngeal carcinoma cells, fisetin regulates ERK phosphorylation rather than expression (29), and fisetin promotes the phosphorylation of ERK in cholangiocarcinoma cells (30). In the current study, fisetin did not reduce the levels of phosphorylated ERK in pancreatic cancer cells.
The results of the present study showed that fisetin down-regulated ERK1 at both protein and transcriptional levels, whereas it down-regulated ERK2 only at the protein level. We speculate that fisetin specifically interacts with transcription factors or repressors of ERK1/2 genes in pancreatic cancer. Although ERK1 and ERK2 share 84% amino acid sequence homology, knocking out ERK1 or ERK2 in mice produces different phenotypes, suggesting that these isoforms have distinct functions (31). Thus, fisetin treatment may affect the distinct functions of these isoforms.
In conclusion, this study suggests a potential application for combination treatment with fisetin and gemcitabine to enhance cytotoxicity in MiaPaca-2 cells. This is important for the early treatment of pancreatic cancer, which shows resistance to gemcitabine. These observations indicate that the combination of fisetin and gemcitabine may be beneficial to eliminate pancreatic cancer and inhibit recurrence.
Acknowledgements
This study was supported by a grant of the Korean Society of Gastrointestinal Cancer (2014-2) for 2014. Nayoung Kim was supported by a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (Grant Number: HI15C0972).
Footnotes
↵* These Authors contributed equally to this study.
Conflicts of Interests
None.
- Received April 17, 2018.
- Revision received May 14, 2018.
- Accepted May 15, 2018.
- Copyright© 2018, International Institute of Anticancer Research (Dr. George J. Delinasios), All rights reserved
References
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.