


Abstract
Background/Aim: Anti-cancer activity of 3,5,7-trihydroxyflavone (galangin) has been documented in a variety of cancer types; however, its effect on human nasopharyngeal carcinoma (NPC) cells remains undetermined. Materials and Methods: Human NPC cell lines were treated with galangin. Apoptosis was analyzed by assessing nuclear condensation, cleavage of pro-caspase-3 and poly ADP-ribose polymerase (PARP), and DNA fragmentation. Short hairpin RNA-mediated silencing of p53 was used for characterizing the role of p53 in the anti-cancer activity of galangin. Phosphatidylinositol 3-kinase (PI3K) inhibitor, protein kinase B (AKT) inhibitor, and ectopic expression of wild type p85α or p85α mutant lacking p110α-binding ability were utilized to confirm the involvement of PI3K/AKT inactivation in galangin-induced apoptosis. Results: Galangin induces apoptosis and S-phase arrest by attenuating the PI3K/AKT signaling pathway. Silencing of p53 did not block the anti-cancer activity of galangin on NPC cells. Conclusion: Galangin effects on apoptosis and S-phase arrest in NPC cells are mediated via interfering with the PI3K-AKT signaling pathway in a p53-independent manner.
Nasopharyngeal carcinoma (NPC) arising from the epithelium of the nasopharynx is one of the most common malignancies in Southeast Asian populations, especially among Chinese people (1, 2). Clinically, this cancer exhibits a high incidence of lymph node spread and distant metastasis that contribute to its poor prognosis (3). Aberrant activation of phosphatidylinositol 3-kinase (PI3K)/protein kinase B (AKT) signaling pathway has been reported to associate with early metastatic disease progression, poor clinical outcome, and chemotherapy resistance in NPC patients (4). AKT, a downstream target of PI3K, can be hyperactivated by overexpression of the p110α catalytic subunit of PI3K, which plays a key role in lipid signaling pathway of malignant progression (5). Constitutively activated PI3K–AKT signaling up-regulates the expression of survival and oncogenic genes, which protect cancer cells against apoptosis and promote their proliferation and invasion (6, 7). Among the B-cell lymphoma 2 (BCL-2) family proteins, BCL-2 and BCL-xL are suggested as important anti-apoptotic proteins, up-regulated by AKT signaling (8, 9). Pro-apoptotic activation and mitochondrial oligomerization of BAX and BAK can also be negatively regulated by activated AKT (10, 11). Involvement of anti-apoptotic BCL-2/BCL-xL and apoptotic BAX/BAK proteins in the regulation of cell survival is documented by modulating the mitochondrial release of cytochrome c (Cyt c) (12). Upon apoptotic stimuli, Cyt c is released from the mitochondrial intermembrane space into the cytosol and then triggers the formation of an apoptosome composed of apoptotic protease-activating factor 1 (APAF-1), dATP, caspase-9, and Cyt c. Apoptosome formation leads to activation of caspase-9, which, subsequently, causes activation of the downstream effector caspases-3, thereby executing the apoptotic cell death (13).
The cross-regulation between PI3K–AKT signaling and wild-type p53 in cell survival is well-documented and extensively studied (14). Mutant p53 gain-of-function was found to associate with elevated AKT activity, which has been suggested to correlate with poor prognosis or metastasis in cancer patients (15-18). However, loss of AKT activity has been reported to promote apoptosis of cancer cells in a p53-independent manner (19-25). p53-knockout or deficiency render cancer cells sensitive to a series of apoptosis inducers (19, 20, 22, 24). There is increasing evidence that p53-independent apoptosis is induced by enhanced activity of caspase-3 and cleavage of the caspase substrate poly (ADP-ribose) polymerase (PARP), mediated by caspase-9 (26-28). Several p53 mutants have dominant–negative effects that exert oncogenic gain of function through activation of growth factor receptor (17, 29-34). Thus, interaction between mutant p53 and PI3K–AKT signaling in modulating tumorigenesis remains an open question.
The compound 3,5,7-Trihydroxyflavone (galangin) is a naturally-occurring bioflavonoid isolated from Alpinia galangal (35). It has been shown to modulate the function of intracellular anti-apoptotic, apoptotic, and signal proteins and the expression of BCR-ABL oncogene to facilitate apoptosis and growth inhibition of human cancer cells (36-45). Although the cytotoxic effects of galangin on different human cancer cells have been studied, the effects and underlying mechanisms of galangin on human NPC cells remain undetermined. The present study intended to investigate the apoptosis-inducing effect of galangin on human NPC cells and its related mechanism.
Materials and Methods
Cell culture. Human NPC cell lines (NPC-TW 039 and NPC-TW 076) with a G→C mutation at condon 280 causing an arginine to threonine amino acid change were derived from a 64-year-old male and 36-year-old female Chinese patient with keratinizing squamous cell carcinoma (WHO type I), respectively. Both cell lines were obtained as previously described (46) and were cultured routinely in Dulbecco's modified Eagle's medium (DMEM) supplemented with 5% fetal bovine serum (FBS) (both from Gibco BRL. Grand Island, NY, USA). Normal human embryonic lung fibroblast WI-38 cell line expressing wild-type p53 was obtained from the Food Industry Research and Development Institute (Hsinchu, Taiwan). WI-38 cells were cultured in minimum essential medium (MEM) (Gibco BRL. Grand Island, NY, USA). Supplemented with 5% FBS. All cell lines were grown in 10-cm tissue culture dishes at 37°C in a humidified incubator containing 5% CO2.
Chemicals and reagents. PI3K inhibitor, 2-(4-morpholinyl)-8-phenyl-4H-1- benzopyran- 4-one (LY294002), was purchased from Cell Signaling (Beverly, MA, USA). AKT inhibitor, 8-[4-(1-aminocyclobutyl)phenyl]-9-phenyl-1, 2, 4-triazolo[3,4-f][1,6]naphthyridin-3(2H)-onehydrochloride (MK-2206), was obtained from Selleckchem (Houston, TX, USA).
Cell proliferation assay. Cell proliferation was determined by the MTT method. NPC cells were treated with dimethyl sulfoxide (DMSO, vehicle) (Merck, Darmstadt, Germany) or with galangin (Sigma-Aldrich. St. Louis, MO, USA) diluted in DMSO, at concentrations between 10-100 μM for 36 and 48 h. Treated cells were washed once with phosphate-buffered saline (PBS) and incubated with 0.5 mg/ml MTT (Sigma-Aldrich, St. Louis, MO, USA) at 37°C for 5 h. The resulting formazan precipitate was dissolved in 150 μl of DMSO, and the optical density (OD) was determined using an enzyme-linked immunosorbent assay (ELISA) plate reader (Thermo Labsystems Multiskan Spectrum, Waltham, MA, USA) at 570 nm (47).
Measurement of DNA fragmentation. Histone-associated DNA fragments (mono-, oligo- nucleosomes) were measured using a Cell Death Detection ELISA kit (Roche Applied Science, Mannheim, Germany). Briefly, vehicle- or galangin-treated cells were incubated in hypertonic buffer for 30 min at room temperature. After centrifugation, the cell lysates were transferred to an anti-histone-coated microplate to bind histone-associated DNA fragments. The plates were washed after 1.5 h of incubation, and nonspecific binding sites were saturated with blocking buffer. The plates were then incubated with peroxidase-conjugated anti-DNA antibodies for 1.5 h at room temperature. To determine the amount of retained peroxidase, 2,2’-azino-di-(3-ethylbenzthiazoline-6-sulfonate) was added as a substrate, and a spectrophotometer (Thermo Labsystems Multiskan Spectrum, Franklin, MA, USA) was used to measure the absorbance at 405 nm (48, 49).
Plasmid transfection. Cells (at 60-70% confluence in a 12-well plate) were transfected with the HA epitope-tagged wt p85α (HA-wt p85α), FLAG epitope-tagged Δp85α (FLAG-Δp85α), p53 short hairpin RNA (p53 shRNA), or GFP shRNA (Addgene, Cambridge, MA, USA) and expression plasmid using Lipofectamine 2000 (Thermo Fisher Scientific, Waltham, MA, USA). The expression of HA-wt p85α, FLAG-Δp85α, and p53 shRNA in the transfected cells was assessed by Western blot using antibodies specific to PI3K p85α (Santa Cruz Biotechnology, Santa Cruz, CA, USA) and p53 (Cell Signaling, Beverly, MA, USA) (11).
Western blot analysis. Treated or transfected cells were lysed in lysis buffer (50 mM Tris-HCl [pH 8.0], 120 mM NaCl, 1 μg/ml aprotinin, 100 mM Na3VO4, 50 mM NaF, and 0.5% NP-40 (Sigma-Aldrich. St. Louis, MO, USA)). Protein concentration was determined using the Bradford method (Bio-Rad, Hercules, CA, USA). Proteins (30 μg per lane) were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and then transferred to a polyvinylidene difluoride membrane (Immobilon-P; Millipore. Bedford, MA, USA). The membranes were blocked with PBS containing 3% skim milk overnight at 4°C and then incubated with primary antibodies against caspase-3, caspase-9, phospho (p)-AKT (Ser 473), AKT, BAK, p53 and p21 (Cell Signaling, Beverly, MA, USA), BAD and BCL-xL (BD Pharmingen, San Diego, CA, USA), BAX (Upstate Biotechnology, Lake Placid, NY, USA) and BCL-2 (Millipore, Billerica, MA, USA), cytochrome c (Cyt c), PARP and p85α (Santa Cruz Biotechnology, Santa Cruz. CA, USA), γ-tubulin (Sigma-Aldrich, St. Louis, MO, USA), overnight 4°C. The proteins were labeled using horseradish peroxidase-conjugated goat anti-mouse, goat anti-rabbit, or donkey anti-goat IgG secondary antibodies (Jackson ImmunoResearch Laboratories, West Grove, PA, USA), and visualized by chemiluminesence with Western Blotting Luminol Reagent (Santa Cruz Biotechnology, Santa Cruz, CA, USA) (48).
Measurement of cell cycle by flow cytometry. Cells (1×105) were trypsinized, washed twice with PBS, and fixed in 80% ethanol. Fixed cells were washed with PBS, incubated with 100 μg/ml RNase for 30 min at 37°C, stained with propidium iodide (PI) 50 μg/ml (Sigma-Aldrich. St. Louis, MO, USA), and analyzed using a FACSCount flow cytometer (BD biosciences, San Jose, CA, USA). The percentage of cells that had undergone apoptosis was calculated as the ratio of the fluorescent area that was smaller than the G0-G1 peak to the total fluorescent area (50).
DAPI staining for the analysis of nuclear morphological changes. Cells were seeded at a density of 1×105 cells per well into 12-well plates. After 16 h of incubation, cells were cultured with vehicle or galangin for 36 h at 37°C. Treated cells were fixed with 4% paraformaldehyde for 20 min, permeabilized with 0.1% Triton X-100 (Sigma-Aldrich, St. Louis, MO, USA) and stained with 1 μg/ml of DAPI (Sigma-Aldrich, St. Louis, MO, USA) for 10 min at RT. Cells were then washed twice with PBS. The morphological changes were observed using an inverted phase contrast and fluorescence microscope (Nikon, Tokyo, Japan) (11).
Establishment of cell clones that stably express p53 shRNA or GFP shRNA. To establish cells stably expressing p53 shRNA or GFP shRNA, cells were transfected using Lipofectamine 2000 (Thermo Fisher Scientific. Waltham, MA, USA) with pPuro-p53 shRNA or pPuro-GFP shRNA plasmid. The transfected cells were selected and cloned in the presence of 2 μg/ml puromycin. The efficiency of p53 knockdown was confirmed by western blot analysis with an anti-p53 antibody, as described above (47).
Statistical analysis. Statistical analysis of the data was performed using an unpaired Student's t-test and ANOVA. The data from at least three independent experiments were presented as mean±SD and were considered statistically significant when p<0.05.
Results
Induced apoptosis in galangin-treated NPC cells was associated with caspase-3 activation and PARP cleavage. To assess the cytotoxic effect of galangin on the human NPC and normal human fibroblast cells, cells were exposed to concentration ranging from 10 μM to 100 μM of galangin for 36 and 48 h. Under the culture conditions, galangin effectively showed dose-dependent growth inhibition of NPC-TW 039 and NPC-TW 076 cells, with half-maximal inhibitory concentration (IC50) value of 60 μM. The ODs (570 nm) of NPC-TW 039 and 076 cells treated with 60 μM of galangin for 36 h were 1.407±0.19 (p<0.05) and 1.308±0.072 (p<0.05), respectively, compared with the corresponding vehicle control ODs 3.675±0.032 and 2.442±0.113. Cells treated for 48 h showed ODs of 1.434±0.102 (p<0.05) and 1.083±0.024 (p<0.05), compared with the corresponding vehicle control ODs of 3.830±0.076 and 2.965±0.163, respectively (Figure 1A). Increased numbers of apoptotic bodies and condensed/fragmented nuclei were observed in both NPC-TW 039 and NPC-TW 076 cell lines treated with 60 μM of galangin for 36 and 48 h (Figure 1B and 1C). Although galangin exhibited a growth-inhibitory effect on normal human fibroblast WI-38 at concentrations ranging from 10 μM to 100 μM, at a concentration of 60 μM it did not alter the morphology and nuclear condensation/fragmentation of cells (Figure 1).
To examine whether induction of growth inhibition by galangin could be linked to apoptosis and caspase activation, apoptotic DNA fragmentation was assessed by detection of histone-associated DNA fragments and caspase-3 activation was determined by western blot analysis. Cleavage of pro-caspase-3 and PARP occurred in galangin (60 μM)-treated NPC cells but not in galangin (60 μM)-treated WI-38 cells (Figure 2A). Significant increase in DNA fragmentation was detected in NPC-TW 039 and NPC-TW 076 cell lines treated with galangin for 36 h (OD405: 0.973±0.072, p<0.05 and 0.810±0.084, p<0.05, respectively, compared to vehicle control 0.133±0.020 and 0.186±0.054) and 48 h (OD405: 1.252±0.033, p<0.05 and 1.20±0.009, p<0.05, respectively, compared with vehicle control 0.159±0.017 and 0.176±0.024). However, treatment of WI-38 cells with galangin also resulted in increased level of DNA fragmentation (Figure 2B). Thus, concentration of 60 μM was used to treat cells in all subsequent experiments. These results suggest that galangin induced caspase-3 activation followed by apoptosis in NPC cells.
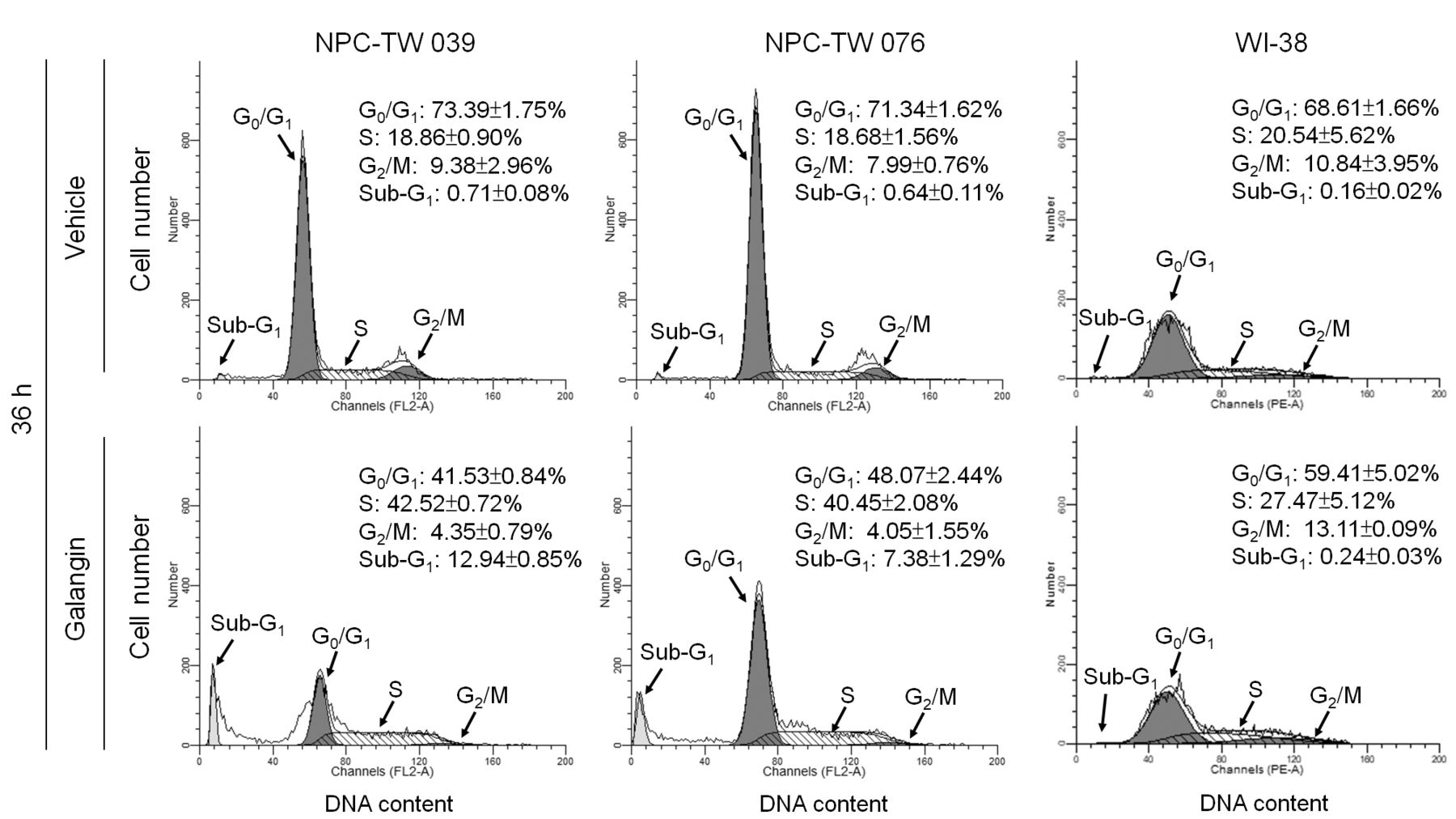
Retardation of NPC cells in the S-phase of the cell cycle, induced by galangin. To investigate whether a reduced growth of NPC cells impaired progression of cell cycle, we tried to analyze the cell cycle through measuring DNA contents by using flow cytometric analysis. The galangin treatment delayed progression of cell cycle in both NPC-TW 039 and NPC-TW 076 cell lines, and the percentages of cells in S-phase were similar (42.52±0.72% and 40.45±2.08%, respectively). A significant increase in the number of apoptotic cells (sub-G1-phase population) was also observed in galangin-treated NPC-TW 039 and 076 cells compared to vehicle-treated control cells (0.71±0.08% vs.12.94±0.85%, p<0.05 and 0.64±0.11% vs. 7.38±1.29%, p<0.05, respectively). However, galangin treatment did not cause S-phase arrest and increase in the sub–G1-phase population in the WI-38 cells (Figure 3). These results indicate that galangin induced apoptosis and caused an arrest of cell-cycle in S-phase in NPC cells.
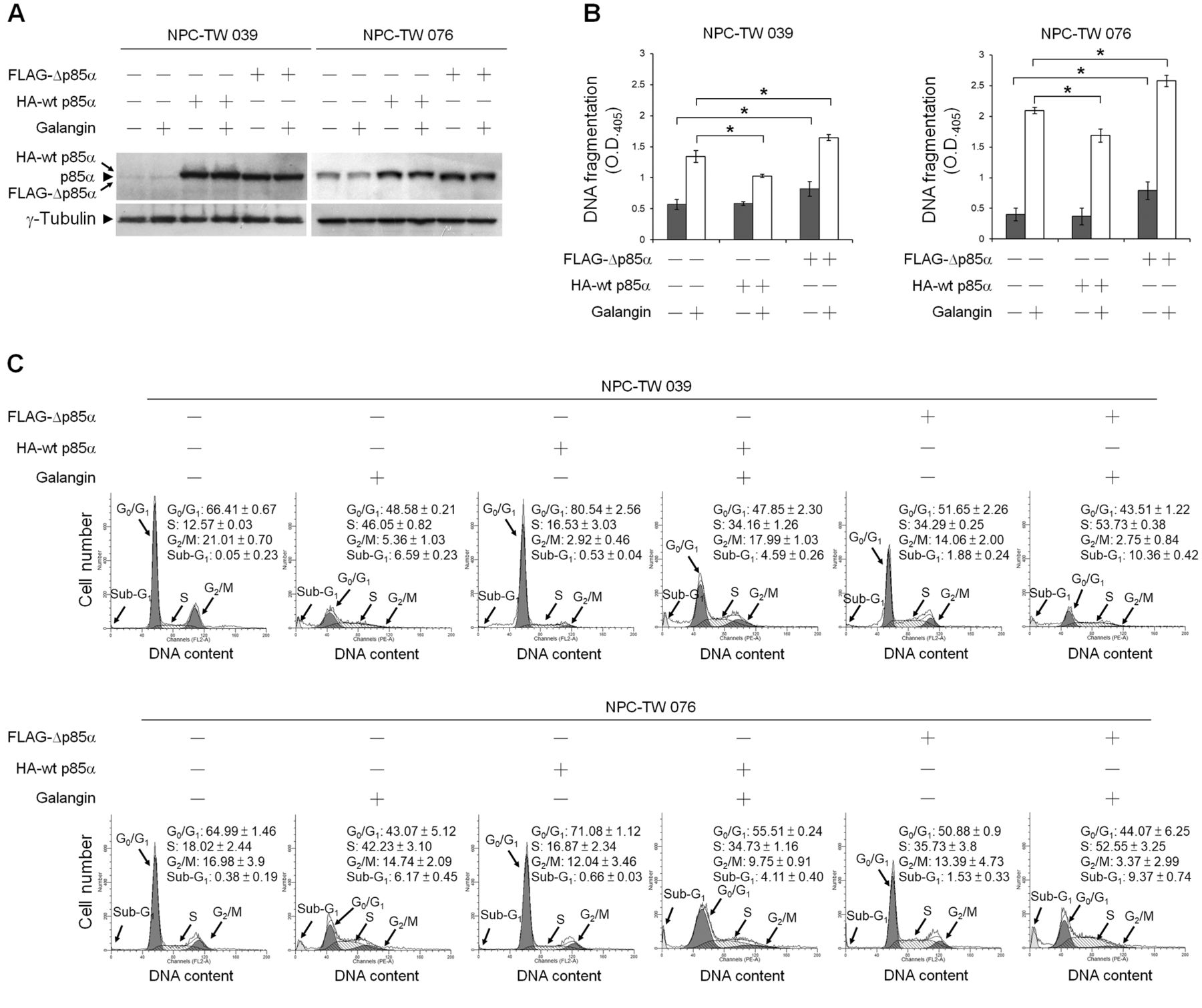
Suppression of PI3K–AKT Signaling involved in galangin-induced NPC cell apoptosis. Western blot analysis with an anti-phospho (p)-AKT (Ser 473) antibody revealed that reduced levels of p-AKT (Ser 473) were detected in whole-cell lysates of galangin-treated NPC and WI-38 cells (Figure 4B). To verify the importance of PI3K–AKT signaling in cell survival, cells were co-treated with an inhibitor of PI3K or AKT. Figure 4A shows that addition of the PI3K inhibitor (LY294002) or AKT inhibitor (MK-2206) induced apoptotic morphological changes in NPC cells but not in WI-38 cells. When NPC cells were co-treated with galangin and LY294002 or galangin and MK-2206, induction of apoptotic morphological changes in NPC cells was observed. Western blot analysis showed that inhibition of PI3K with LY294002 or inhibition of AKT with MK-2206 was able to induce the cleavage of pro-caspase-9, pro-caspase-3, and PARP in NPC cells. While co-treatment of NPC cells with galangin and LY294002 or galangin and MK-2206 effectively increased the cleavage of pro-caspase-9, pro-caspase-3, and PARP (Figure 4B). To confirm that inhibition of PI3K–AKT signaling is required for galangin-induced apoptosis in NPC cells, the transient ectopic expression of wild-type p85α (wt p85α) or mutant p85α (which lacks the binding site for the p110 catalytic subunit of PI3K; Δp85α) was used. Overexpression of wt p85α in NPC-TW 039 and 076 cells treated with vehicle did not significantly alter the levels of DNA fragmentation and the percentage of S-phase fraction, compared to vehicle-treated control vector-transfected cells. In contrast, ectopic expression of Δp85α in vehicle-treated NPC-TW 039 and 076 cells elevated both DNA fragmentation (0.820±0.117 vs. 0.571±0.081, p<0.05 and 0.787±0.146 vs. 0.399±0.101, p<0.05, respectively) and the percentage of S-phase-arrested cells (34.29±0.25% vs. 12.57±0.03, p<0.05 and 35.73±3.8% vs. 18.02±2.44, p<0.05, respectively) compared to vehicle-treated control vector-transfected cells.
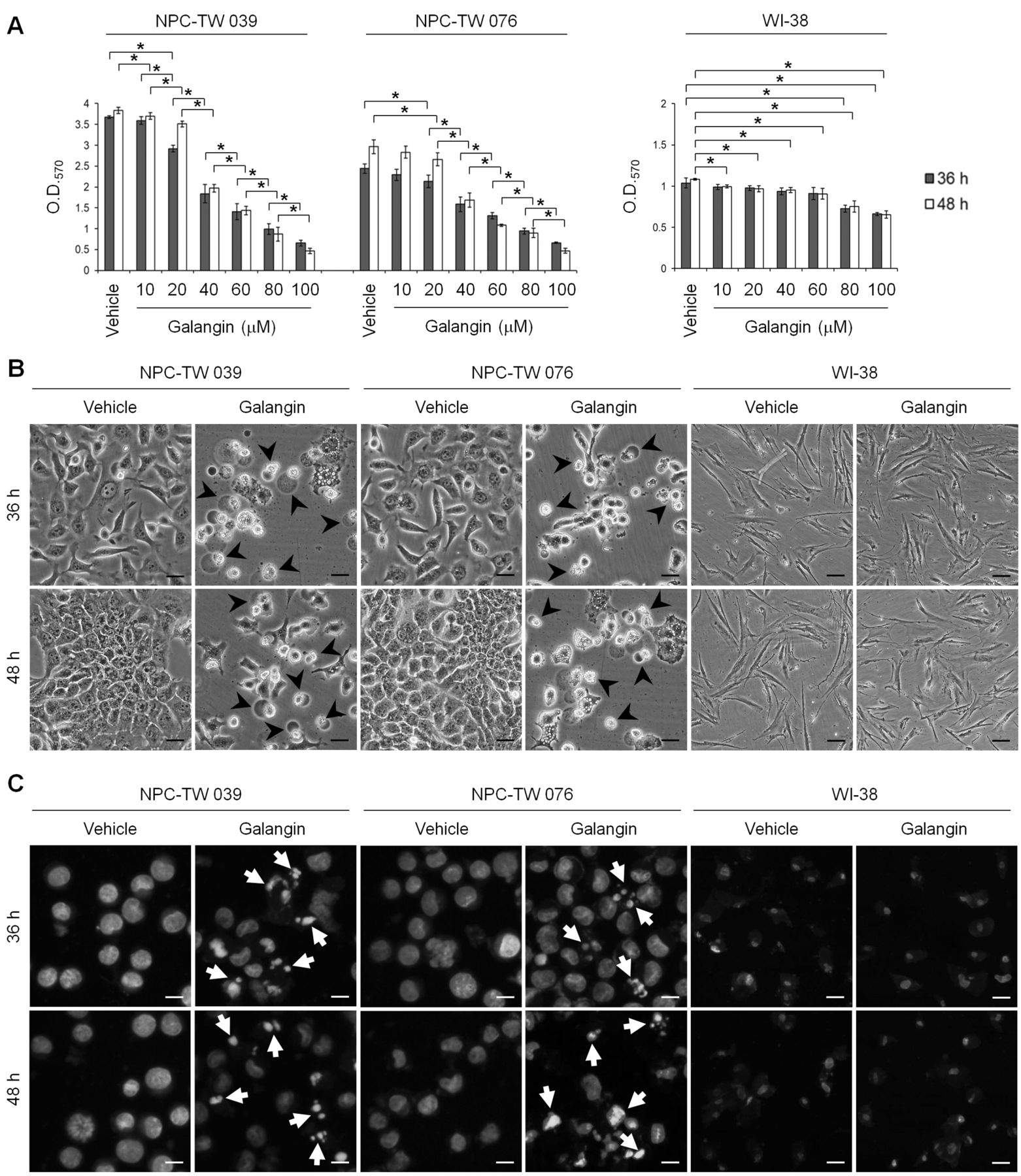
The growth-inhibitory and apoptotic effects of galangin on human nasopharyngeal carcinoma (NPC) cells. (A) The human NPC and normal human fibroblast WI-38 cells were treated with the indicated concentrations of galangin for 36 and 48 h. Cell proliferation was determined by MTT. The values presented are the mean±standard error from three independent experiments. *p<0.05: significantly different from vehicle (−)-treated cells or galangin-treated cells. (B and C) Cells were treated for 36 and 48 h with vehicle or galangin (60 μM). Cell morphology and DAPI stained nuclei were examined using an inverted phase-contrast microscope and an inverted fluorescence microscope, respectively. Photographs were taken under ×150 inverted phase-contrast light microscopy, scale bar, 50 μm (B) and ×200 inverted fluorescence microscope, scale bar, 50 μm (C). The arrowheads and arrows indicate apoptotic cells and condensed nuclei, respectively.

Effects of galangin on the induction of pro-caspase-3 cleavage, poly (ADP-ribose) polymerase (PARP) cleavage, and DNA fragmentation in human nasopharyngeal carcinoma (NPC) cells. Human NPC and normal human fibroblast WI-38 cells were treated for 36 h with vehicle or galangin (60 μM). (A) The protein levels of caspase-3 and PARP in cell lysates were analyzed using specific antibodies. γ-Tubulin was used as an internal control for sample loading. (B) DNA fragmentation was determined using a Cell Death Detection ELISA kit. Values are presented as the mean±standard error from three independent experiments. *p<0.05: statistically significant differences compared to vehicle-treated cells.
On the other hand, when compared galangin-treated, wt p85α-tranfected NPC-TW cells to the control vector-transfected group, ectopic expression of wt p85α was shown to significantly inhibit galangin-induced DNA fragmentation (1.029±0.02 vs. 1.343±0.09, p<0.05 for NPC-TW 039 and 1.686±0.107 vs. 2.092±0.05, p<0.05 for NPC-TW 076), which simultaneously resulted in decreased percentage of S-phase-arrested cells (34.16±1.26% vs. 46.05±0.82%, p<0.05 for NPC-TW 039 and 34.73±1.16% vs. 42.23±3.10%, p<0.05 for NPC-TW 076). However, Δp85α overexpression enhanced the levels of galangin-induced DNA fragmentation (1.647±0.045 vs. 1.343±0.09, p<0.05 for NPC-TW 039 and 2.577±0.09 vs. 2.092±0.05, p<0.05 for NPC-TW 076) and the percentage of S-phase-arrested cells (53.73±0.38% vs. 46.05±0.82%, p<0.05 for NPC-TW 039 and 52.55±3.25% vs. 42.23±3.10%, p<0.05 for NPC-TW 076) compared to galangin-treated control vector-transfected, or compared to galangin-treated wt p85-transfected NPC cells (DNA fragmentation: 1.647±0.045 vs. 1.029±0.02, p<0.05 for NPC-TW 039 and 2.577±0.09 vs. 1.686±0.107, p<0.05 for NPC-TW 076; percentage of S-arrested cells: 53.73±0.38% vs. 34.16±1.26%, p<0.05 for NPC-TW 039 and 52.55±3.25% vs. 34.73±1.16%, p<0.05 for NPC-TW 076) (Figure 5). These results indicate that galangin induced apoptosis and S-phase arrest of NPC cells by suppressing PI3K-AKT signaling pathway.
Galangin inhibited cell survival mediated by PI3K-AKT signaling and caused S-phase arrest in a p53-independent manner in NPC cells. The regulation of caspase-3-mediated apoptosis by the PI3K-AKT pathway has been reported to occur via downstream targets, p53 and the BCL-2 family proteins (8-11, 15). To verify the role of p53 in the galangin-induced apoptotic response, we generated a panel of p53 shRNA-transfected NPC-TW 039 and 076 cell clones. Three independent clones from each experimental group along with a clone stably expressing the GFP shRNA (control cells) were chosen for further study. As shown in Figure 6A, cells stably expressing the p53 shRNA clones had reduced (p53: 9.36±0.94 vs. 2.16±0.45 [p<0.05] for K4 cells, 11.29±2.05 vs. 3.02±1.05 [p<0.05] K5 cells, respectively) and undetectable (K9, K17, K1 and K2 clones) levels of p53 protein by galangin-treated p53 shRNA NPC clone cells induction, compared with the galangin–treated control GFP shRNA NPC clone cells. Galangin treatment increased the levels of p21, BAX, BAD, and BAK protein expression in GFP shRNA NPC cells. Moreover, galangin treatment decreased BCL-2 and BCL-xL protein levels in GFP shRNA NPC cells (Figure 6A and Table I). Interestingly, galangin increased DNA fragmentation level in 2 of the 3 clones of p53 shRNA-expressing NPC-TW 039 and 076 cells (K9: 2.12±0.14 vs. 1.24±0.07, p<0.05; K17: 2.36±0.13 vs. 1.24±0.07, p<0.05; K1: 2.54±0.07 vs. 1.96±0.11, p<0.05; K2: 2.83±0.16 vs. 1.96±0.11 p<0.05), compared to galangin-treated GFP shRNA-expressing control cells (Figure 6B). To explore whether p53 activity is responsible for cell growth inhibition by galangin, flow cytometry was used to determine the effect of galangin on the cell cycle profile. The percentage of S-phase arrested cells was increased in galangin-treated compared to vehicle-treated p53 shRNA-expressing cells (38.92±0.34% vs. 15.58±1.72%, p<0.05 for NPC-TW 039 and 38.26±4.22% vs. 19.31±0.95%, p<0.05 for NPC-TW 076), as well as in galangin-treated compared to vehicle-treated GFP shRNA-expressing cells (36.25±1.11% vs. 13.76±0.72%, p<0.05 for NPC-TW 039 and 36.89±3.02% vs. 16.04±1.81%, p<0.05 for NPC-TW 076). A significant increase in the number of sub–G1-phase population was also observed in galangin-treated compared to vehicle-treated p53 shRNA-expressing cells (10.88±0.34% vs. 0.60±0.01%, p<0.05 for NPC-TW 039 and 10.54±2.05% vs. 0.31±0.01%, p<0.05 for NPC-TW 076), as well as in galangin-treated compared to vehicle-treated GFP shRNA-expressing cells (5.35±1.35% vs. 0.59±0.01%, p<0.05 for NPC-TW 039 and 5.36±1.54% vs. 0.42±0.21%, p<0.05 for NPC-TW 076) (Figure 7). These results clearly indicated that induction of both apoptotic cell death and S-phase arrest by galangin occurs independently of p53 expression.
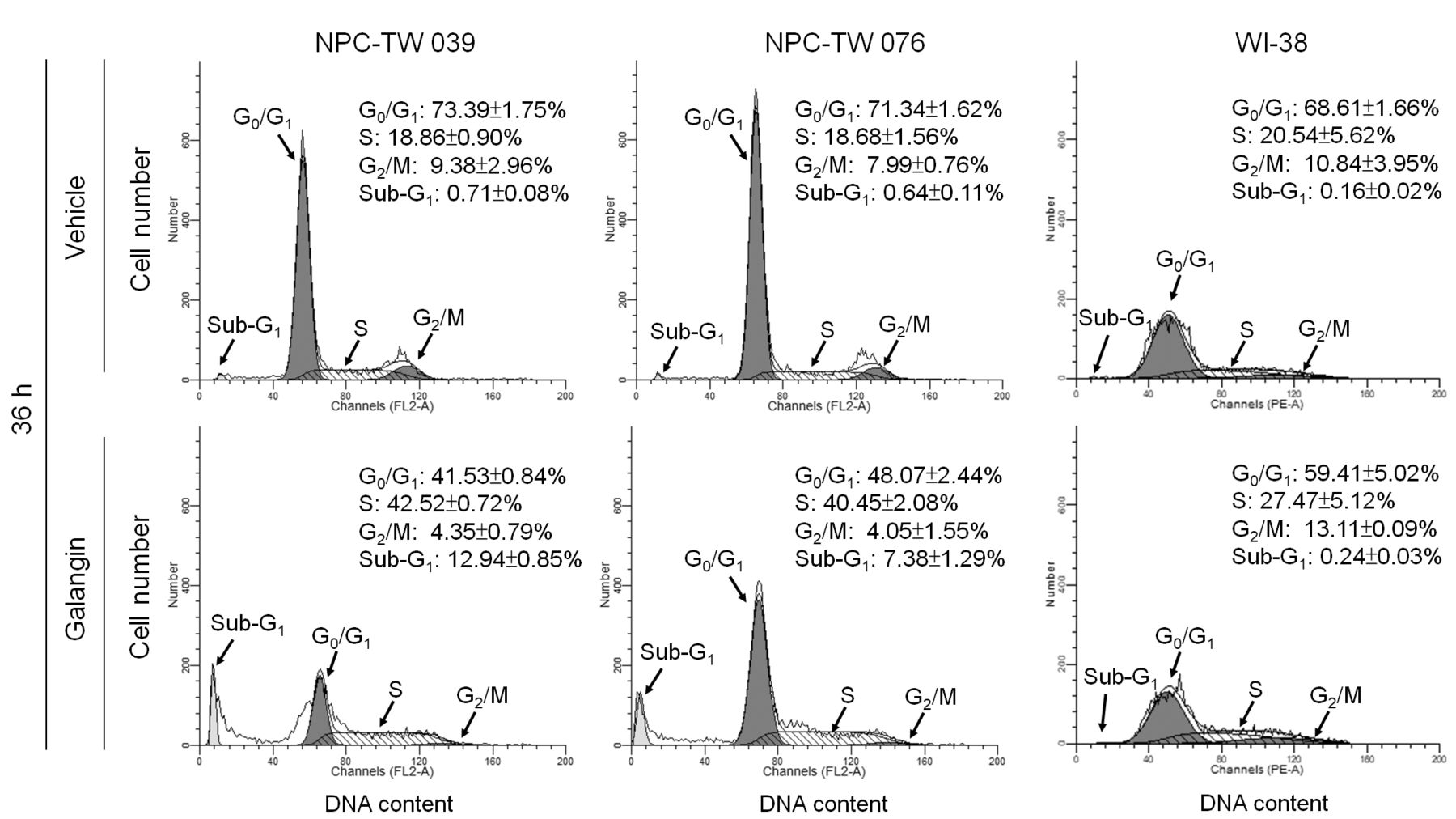
Effect of galangin on cell-cycle progression of human nasopharyngeal carcinoma (NPC) cells. The human NPC and normal human fibroblast WI-38 cells were treated for 36 h with vehicle or galangin (60 μM). When treatment with galangin for 36 h in NPC cells could elevates sub-G1 phase and S phase of percentage and reduces G0/G1 phase but in WI-38 not. The cell-cycle profile was analyzed by flow-cytometric analysis of propidium iodide-stained cells. The values presented are the mean±standard error from three independent experiments.

Effects of galangin, LY294002, and MK-2206 on apoptosis induction in human nasopharyngeal carcinoma (NPC) cells. (A and B) The human NPC and normal human fibroblast WI-38 cells were treated for 36 h with vehicle, galangin (60 μM), LY294002 (10 μM), or MK-2206 (100 nM). (A) Cell morphology was examined using an inverted phase-contrast microscope. Photographs were taken under ×150 inverted phase-contrast light microscopy, scale bar, 50 μm. (B) The levels of p-protein kinase B (AKT) (Ser 473), AKT, p53, pro-caspase-9, pro-caspase-3, and poly (ADP-ribose) polymerase (PARP) in the treated-cell lysates were analyzed using specific antibodies. γ-Tubulin was used as an internal control for sample loading.
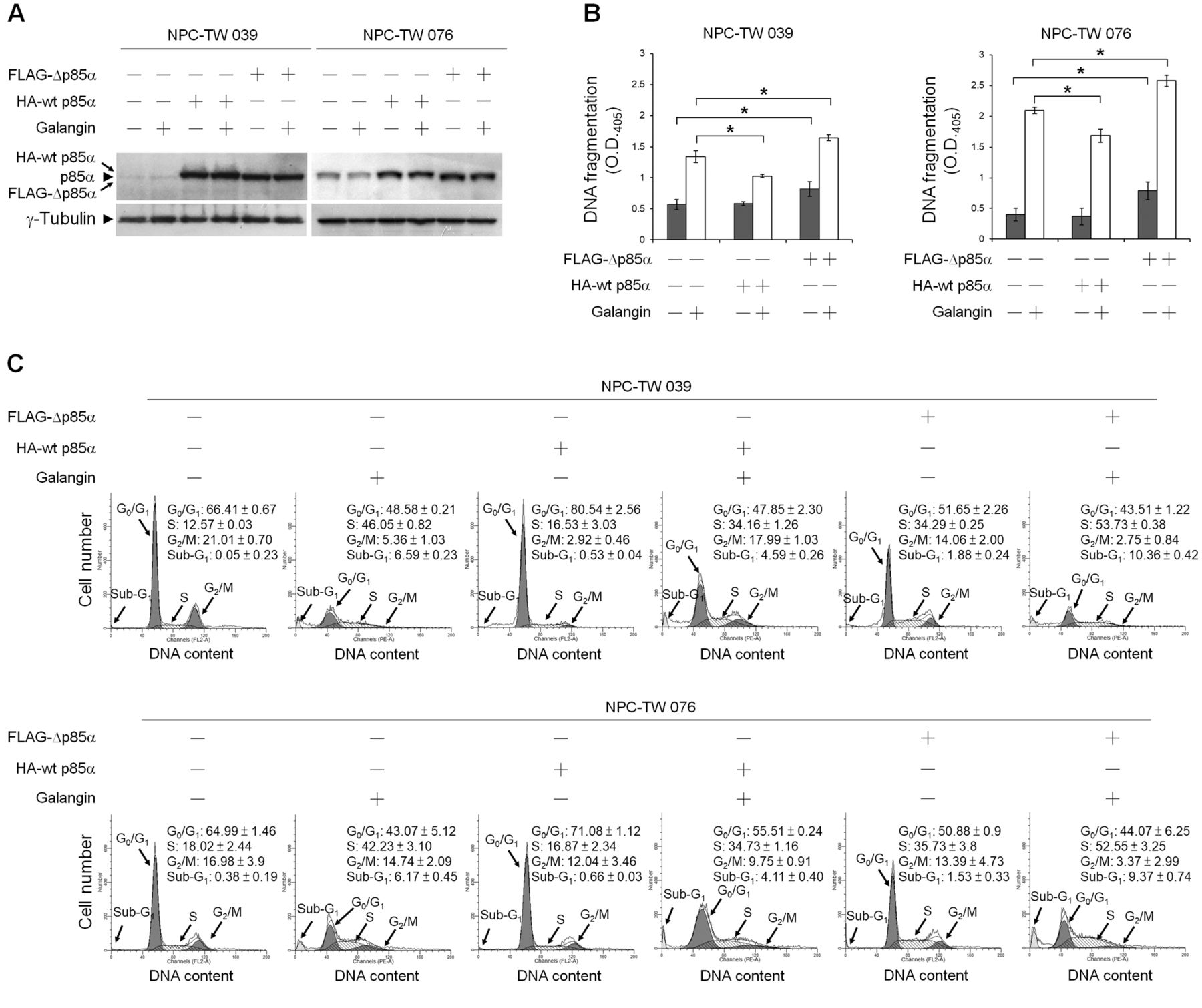
Ectopic expression of wild-type p85α overcomes cell-cycle S-phase arrest induced by galangin. At 12 h after transfection with wild-type p85α (wt p85α) or p85α mutant lacking p110α-binding site (Δp85α), human nasopharyngeal carcinoma (NPC) cells were treated with either vehicle or galangin (60 μM) for 36 h. (A) The levels of HA-wt p85α, FLAG-Δp85α, and Δp85α in the treated-transfected cell lysates were analyzed using specific anti-p85α antibody. γ-Tubulin was used as an internal control for sample loading. (B) DNA fragmentation was determined using a Cell Death Detection ELISA kit. (C) The cell-cycle profile was analyzed by flow-cytometric analysis of propidium iodide-stained cells. The values presented are the mean±standard error from three independent experiments. *p<0.05: statistically significant differences compared to empty vector-transfected vehicle-treated cells or empty vector-transfected galangin-treated cells.
Discussion
The observed PI3K-AKT inactivation-mediated apoptotic toxicity and S-phase arrest of galangin was selective for human NPC cells but not for normal human fibroblast WI-38. Although galangin induced a decrease in p-AKT (Ser 473) level and a significant increase in DNA fragmentation activity in WI-38 cells, no induction of pro-caspase-3, pro-caspase-9, and PARP cleavage as well as increase of sub–G1-phase and S-phase cell population was observed after treatment with galangin. p85α is predominantly found as a monomer in the cytosol of most types of normal cells (51). In normal cells p85α is expressed at a higher level than p110, and it is thought to be the major effector to modulation normal growth and metabolism of responses to most stimuli (51-53). Luo et al. have shown that monomeric p85α can act as a downstream target of the insulin-like growth factor-1 receptor to transduce survival signal (54). Using ATP-competitive inhibition assay and thin-layer chromatography separation of the γ32P-labeled phosphatidylinositol 3,4,5-triphosphate (PIP3) product of kinase reaction with purified recombinant p85α and p110α proteins, we found that galangin might be an ATP-competitive inhibitor and did inhibit PI3K activity (our unpublished data). Accordingly, we propose that galangin blocks AKT activation by targeting the catalytic subunit of PI3K, p110α, thereby suppressing the signal transduction effect of PI3K-AKT and triggering the apoptosis and cell-cycle arrest at S-phase of NPC cells. The inability of galangin to induce S-phase cell cycle arrest or apoptosis on normal human fibroblast cells might be attributed to the targeting to p110α, which aborts PI3K-AKT signal transduction events, and in turn activates the monomeric p85α-mediated signaling pathway resulting in cell survival (54).

Cells expressing stable short hairpin RNA (shRNA) targeting p53 did not alter the inhibitory effect of galangin on phosphatidylinositol 3-kinase (PI3K) (PI3K)/protein kinase B (AKT)-mediated survival signaling. Green fluorescent protein (GFP) shRNA or p53 shRNA–expressing stable cells were treated with either vehicle or galangin (60 μM) for 36 h. (A) The levels of indicated protein in the treated-cell lysates were analyzed using specific antibodies. γ-Tubulin was used as an internal control for sample loading. (B) DNA fragmentation was determined using a Cell Death Detection ELISA kit. The values presented are the mean±standard error from three independent experiments. *p<0.05: statistically significant differences compared to GFP shRNA vehicle-treated cells or p53 shRNA vehicle-treated cells.
The present study demonstrated the effect of galangin on the growth and cell cycle progress of NPC cells In a previous study it was found that galangin treatment decreased the levels of AKT (Ser 473) phosphorylation and arrested human oral squamous cell carcinoma (OSCC) cells in G0/G1 phase (42). Our findings indicated that the S-phase cell cycle arrest of NPC cells induced by galangin is associated with the suppression of AKT (Ser 473) phosphorylation. It was also shown that ectopic expression of wt p85α, but not p85α mutant (lacking p110α-binding site), allowed NPC cells to overcome the galangin-induced S-phase arrest. Furthermore, it is well-known that the PI3K-AKT pathway is activated during early G1 phase and late S-phase in cells progressing from G1 into G2/M-phase by the phosphorylation of AKT at Ser 473 (55-57). Combined, this evidence suggests that induction of Ser 473 phosphorylation of AKT is involved in the regulation of S-G2/M phase cell cycle progression in NPC cells. Therefore, the galangin-induced S-phase arrest of NPC cells, through PI3K-AKT signaling suppression, appears to be cell type-dependent, probably because of the differential status of the PI3K signaling pathway in various cancer cell lines (58-60).
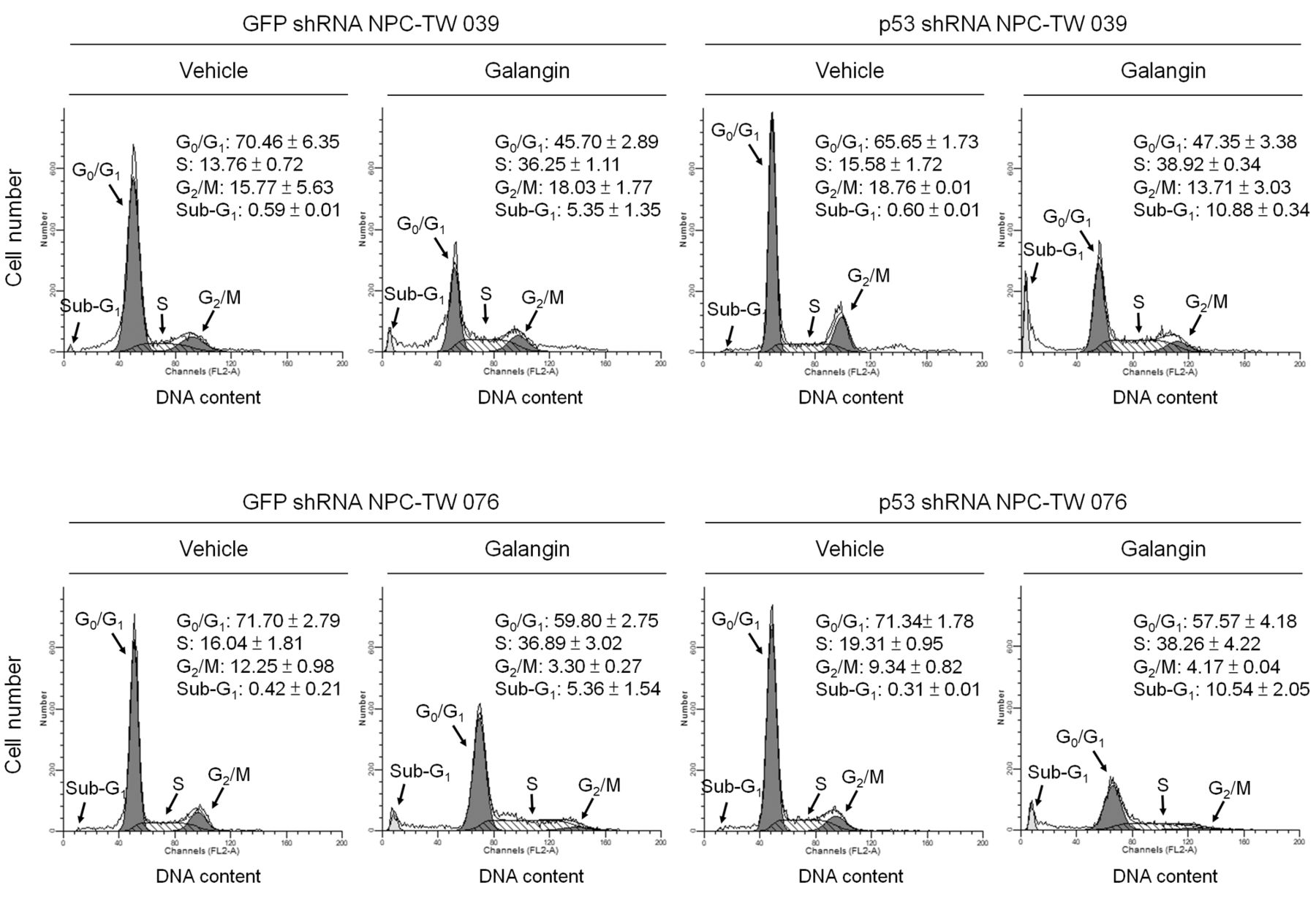
p53 is not involved in the induction of cell-cycle S-phase arrest by galangin. Green fluorescent protein short hairpin RNA (GFP shRNA) or p53 shRNA-expressing stable cells were treated with either vehicle or galangin (60 μM) for 36 h. The cell cycle profile was analyzed by flow-cytometric analysis of propidium iodide-stained cells. The values presented are the mean±standard error from three independent experiments.
The finding showed that p53 expression was up-regulated in galangin-treated NPC cells. The transcription of the p53 gene is increased during the S-phase of cell cycle, allowing the rapid response of the cell to DNA damage (61). Although galangin itself did not directly cause DNA damage (62), the ensuing apoptotic signal induced by PI3K–AKT inhibition results in the activation of caspase-3-mediated PARP cleavage, which is also activated in response to DNA damage (63). In the present study, NPC-TW 039 and NPC-TW 076 cells possess a missense mutation in codon 280 of exon 8 (AGA→ACA) resulting in the generation of mutant p53 (R280T) (our unpublished data). Silencing of p53 expression by shRNA delays cell proliferation (data not shown) and results in an increased sensitivity to galangin in NPC cells, suggesting that R280T mutation confers an oncogenic gain-of-function of p53 providing survival advantage to the NPC cells. The function of mutant p53 (R280T) has been found not to exhibit transactivation activity in nasopharyngeal carcinoma cells CNE2 (64). Consistently, the present data indicated that mutant p53 (R280T) loses its transactivation activity for targeted gene, since silencing of the mutant p53 (R280T) failed to affect the expression level of p21, BAX, and BAK in NPC cells. However, up-regulation of p21, BAX, and BAK expression by galangin was observed upon silencing of mutant p53 (R280T) expression in NPC cells. Inhibition of BCL-2/BCL-xL expression by a specific antisense oligonucleotide can induce p53-independent apoptosis in human C8161 melanoma cells (65). The present study revealed a decrease in BCL-2/BCL-xL levels in galangin-treated p53 shRNA NPC cells. Evidently the study provides novel mechanistic insight into the induction of cell-cycle S-phase arrest and apoptosis in NPC cells by galangin through p53-independent suppression of PI3K-AKT signaling pathway. In summary, the data provide exciting new insights into the therapeutic activity and anti-NPC mechanism of galangin.
Effects of p53 knockdown on the expression of p21, BAX, BAD, BAK, BCL-2, and BCL-XL in galangin-treated NPC cells.
Acknowledgements
S.-S. Chen was supported by a Grant from the Central Taiwan University of Science and Technology (CTU105-P-14), Taiwan.
Footnotes
Conflicts of Interest
The Authors disclose that there are no financial or personal relationships with other people or organizations that could inappropriately influence (bias) the present work.
- Received October 19, 2017.
- Revision received December 25, 2017.
- Accepted January 3, 2018.
- Copyright© 2018, International Institute of Anticancer Research (Dr. George J. Delinasios), All rights reserved





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}