


Abstract
Background/Aim: Breast cancer is the most common malignant cancer type in women, and triple-negative breast cancer (TNBC) is an extremely aggressive subtype of breast cancer with poor prognosis rates. The present study investigated the antitumor effect of polo-like kinase 1 (PLK1) inhibitor in combination with the tankyrase-1 (TNKS1) inhibitor on TNBC cells. Materials and Methods: We evaluated the antitumor effects of combination therapy with PLK1 and TNKS1 inhibitor using cell viability analysis, apoptosis assay and transwell assay for cell invasion and migration in TNBC cells. Results: Combination treatment with PLK1 and TNKS1 inhibitors not only inhibited the invasion and migration capacity of TNBC cells, but also increased the apoptosis and cell death of TNBC cells. The viability of TNBC cells with low expression of β-catenin and high expression of PLK1 was not affected by treatment with PLK1 inhibitor. However, the combination treatment with the TNKS1 inhibitor significantly decreased cell invasion and migration and increased apoptosis. Conclusion: Combination therapy of PLK1 and TNKS1 inhibitors may improve the therapeutic efficacy of the current treatment for TNBC.
Stratification of breast cancer (BC) into distinct histological or molecular subtypes has clinical utility for the prognosis of outcome and prediction of treatment (1, 2). Breast cancer can be categorized by clinical or molecular characteristics as hormone receptor-positive (HR+) or luminal, human epidermal growth factor receptor 2 (HER2)-amplified, and triple-negative (TN) or basal-like. Triple-negative breast cancer (TNBC), a subtype defined as tumors that lack expression of estrogen receptor (ER), progesterone receptor (PR), and HER2, is a highly aggressive disease with poor clinical outcome and limited response to current therapies (3). Studies have suggested that the determination of individual tumor clonal genotypes is required for understanding the biology and therapeutic response of patients with TNBC (4).
The serine/threonine kinase polo-like kinase 1 (PLK1) is known to play an important role in mitosis (5). A striking feature of PLK1 is its localization to numerous subcellular structures during mitosis. Several studies have shown that PLK1 is significantly overexpressed in a wide range of tumors, including breast cancers. Aberrant expression of PLK1 is associated with poor clinical prognosis in cancer patients (6). Furthermore, mutations in PLK1 may contribute to tumor development (7). A growing body of evidence also indicates that depletion of PLK1 prolongs mitotic arrest, promoting apoptotic cell death (8). It is evident that PLK1 is a potential therapeutic target, and that deregulation of PLK1 is an important factor of therapeutic treatment in many cancers, including breast cancer.
The Wnt/β-catenin signaling pathway regulates cell proliferation, migration, and differentiation, making it a major regulator of embryonic and tumor development (9). In the Wnt/β-catenin pathway, axin-1/2 regulates β-catenin stability by inhibition of the poly-ADP-ribosylation activity of tankyrase-1/2 (TNKS). Recent studies have demonstrated that up-regulation of the Wnt/β-catenin pathway in TNBC was associated with metastasis and poor prognosis (10, 11). Moreover, activation of aberrant Wnt/β-catenin signaling is associated with the stemness of cancer stem cells in various breast cancers (12, 13). In addition, a recent review suggested that inhibition of the Wnt/β-catenin signaling pathway may serve as a potential therapeutic target in the treatment of TNBC (14).
In our study, we determined the therapeutic effects of a combination treatment of PLK1 and TNKS1 inhibitors on TNBC cells. Our results indicated that PLK1 was highly expressed in TNBC cells, and the overexpression correlated with the anti-proliferative efficacy of PLK1 inhibitor, GW843682X. In addition, the inhibition of PLK1 by GW843682X decreased the migration and invasion capabilities of TNBC cells by stabilizing TNKS1 protein. Finally, combined treatment with GW843682X and TNKS1 inhibitor, XVA939, significantly reduced invasion and migration and significantly enhanced apoptosis, in comparison to monotherapy in TNBC cells. Therefore, we propose that targeting both PLK1 and TNKS1 with small-molecule inhibitors may be a more effective therapeutic strategy for TNBC chemotherapy.
Materials and Methods
Cell culture. HeLa, MDA-MB231, and MDA-MB468 cells were maintained in Dulbecco's Modified Eagle Medium (DMEM) (HyClone, Logan, UT, USA) containing 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. BT549, MCF7, and SK-BR-3 cells were maintained in RPMI-1640 medium (HyClone, Logan, UT, USA) containing 10% FBS and 1% penicillin/streptomycin. MCF10A cells were maintained in DMEM/DMEM F12 (HyClone, Logan, UT, USA) containing 5% horse serum, 20 ng/ml epidermal growth factor, 0.5 μg/ml hydrocortisone, 100 ng/ml insulin, 100 ng/ml cholera toxin, and 1% penicillin/streptomycin. Cell lines were obtained from the American Type Culture Collection (ATCC; Manassas, VA, USA). Cells were incubated at 37°C in humidified conditions with 5% CO2.
Reagents. The following antibodies were used: anti-PLK1 (F-7), anti-Tankyrase-1/2 (H-350) (Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti-Actin (Sigma, St. Louis, MO, USA), anti-β-catenin, anti-Cleaved PARP (Cell Signaling Technology, Danvers, MA, USA). The following reagents were used: dimethyl sulfoxide (DMSO), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (Sigma St. Louis, MO, USA), XAV939, and GW843682X (Sigma, St. Louis, MO, USA). All plasmid constructs that were used have been previously published (15).
Western blot analysis. Cells were harvested and lysed in nuclear extraction (NE) buffer (20 mM HEPES, pH 7.6; 20% glycerol; 250 mM NaCl; 1.5 mM MgCl2; 0.1% Triton X-100; 1 mM PMSF; 1 mM DTT; and protease inhibitor cocktail). Equal amounts of protein were separated by SDS-PAGE and analyzed by immunoblotting with the indicated antibodies.
Immunofluorescence staining. Cells cultured on coverslips were fixed with 4% paraformaldehyde at 4°C for 30 min. After fixation, cells were washed, permeabilized in phosphate buffered saline (PBS) containing 1% Triton X-100 and 0.5% NP-40, and blocked with 3% bovine serum albumin (BSA) and 1% normal horse serum. Cells were then incubated with primary antibodies, followed by incubation with Alexa Fluor 488- or 594-conjugated secondary antibodies (Invitrogen, Carlsbad, CA, USA). Slides were mounted in a medium containing 4’,6-diamidino-2-phenylindole (DAPI) (Vector Laboratories, Burlingame, CA, USA). Images were acquired on a Zeiss Axiovert 200M microscope, using AxioVision software (Carl Zeiss MicroImaging, Inc. Oberkochen, Germany).
Clonogenic survival assay. Six different breast cancer cell lines were treated with GW843682X at various doses (up to 5 μM). After two to three weeks, cells were fixed with methanol and stained with 0.05% crystal violet (Sigma, St. Louis, MO, USA). Colonies were manually counted, and survival curves were generated from a minimum of three independent experiments.
Transwell migration and invasion assays. Transwell migration assay was performed using uncoated cell culture inserts with 8 μm pores (Corning Inc., Tewksbury, MA, USA). Invasion assay was performed using cell invasion assay kits (Chemicon, Temecula, CA, USA) according to the manufacturer's protocol.
Annexin V/propidium iodide (PI) assay for apoptosis. For Annexin V/propidium iodide (PI) assays, cells were stained with Annexin V–APC and PI and evaluated for apoptosis by flow cytometry, according to the manufacturer's protocol (BD PharMingen, San Diego, CA, USA). Briefly, MDA-MB231 cells (1×106) were washed twice with PBS and stained with 5 μl Annexin V–APC and 10 μl of PI (5 μg/ml) in binding buffer (10 mM HEPES, pH 7.4; 140 mM NaOH; 2.5 mM CaCl2) for 15 min at room temperature in the dark. Apoptotic cells were determined using a Becton-Dickinson FACScan cytofluorometer (Mansfield, MA, USA). Both early apoptotic (Annexin V-positive and PI-negative) and late apoptotic (Annexin V-positive and PI-positive) cells were included in cell death.
TUNEL assay. MDA-MB231 cells (1×104) were cultured on a coverslip and either treated with GW843682X (500 nM), XVA939 (1 μM), or a combination of both inhibitors. Cells were then stained with DAPI or subjected to the TUNEL assay, according to the manufacturer's protocol (Calbiochem, San Diego, CA, USA).
Statistical analysis. All values in this study were reported as mean±standard deviation (SD) and were representative of at least three independent experiments. The student's t-test was used for statistical analyses. A p-value of less than 0.05 was considered statistically significant.
Results
Aberrant expression of PLK1 protein in TNBC cells. We investigated PLK1 gene expression profiles in cancers by searching available data from the Oncomine microarray database (http://www.oncomine.org). We found that PLK1 was significantly overexpressed in a set of 40 invasive breast carcinomas, compared to that in 7 normal breast tissues (p-value=7.9×10−9; fold change=3.671) (16). Interestingly, PLK1 was overexpressed by more than three-fold in a set of 18 triple-negative (HER2/ER/PR-negative) breast carcinomas, compared to that in 19 breast carcinomas tissues with different biomarker status (p<0.005) (16). These data corroborated our findings, indicating that PLK1 levels were significantly overexpressed in triple-negative breast carcinomas (Figure 1A and B). Next, we monitored PLK1 expression levels in six human breast cancer cell lines. As shown in Figure 1C, PLK1 was overexpressed in TNBC cell lines, MDA-MB231, MDA-MB468, and BT549. In contrast, MCF10A, MCF7, and SK-BR-3 cells exhibited lower levels of PLK1. Moreover, immunofluorescent staining revealed that PLK1 expression levels were distinctly higher in MDA-MB-231, MDA-MB468, and BT549 cells compared to those in MCF10A, MCF7, and SK-BR-3 cells (Figure 1D). Next, we investigated the relationship between PLK1 expression and clinicopathological factors of patient survival. Kaplan-Meier analysis revealed that PLK1 expression, as a single variable, significantly affected the survival outcome (Figure 1E). Concurrently high PLK1 expression correlated with significantly shorter overall survival (p<0.001; Figure 1E, left), relapse-free survival (p<0.001; Figure 1E, middle), and relapse-free survival (p-value=0.034; Figure 1E, right) in TNBC patients. Altogether, these observations demonstrated that PLK1 was overexpressed in a significant proportion of breast carcinomas and was linked to poor patient prognosis.
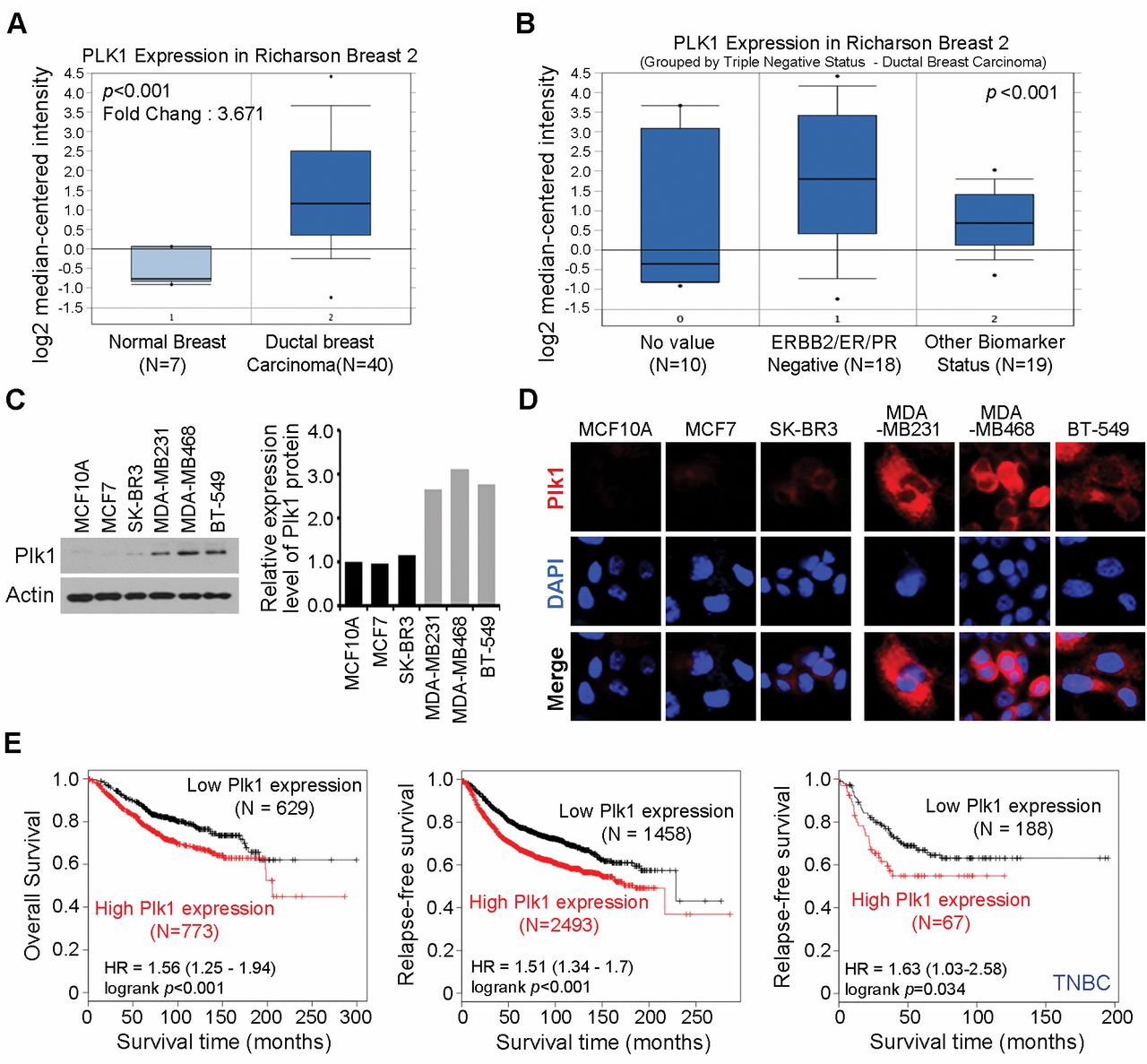
Plk1 was overexpressed in breast cancer. (A and B) Gene expression data were analyzed in Oncomine. The thick bars in the boxes are average expression levels and the boxes represent 95% of the samples. The error bars are above or below the boxes and the range of expression levels is enclosed by two dots. (C) Six different breast Cancer cells were analyzed for Plk1 expression using immunoblotting (left). Pellino-1 expression level was quantified through scanning densitometry with actin as an internal control (right). (D) Immunofluorescence with anti-PlK1 antibody in six different breast cancer cells. (E) Kaplan-Meier survival curve of all breast cancer (left) or triple negative breast cancer patients (right).

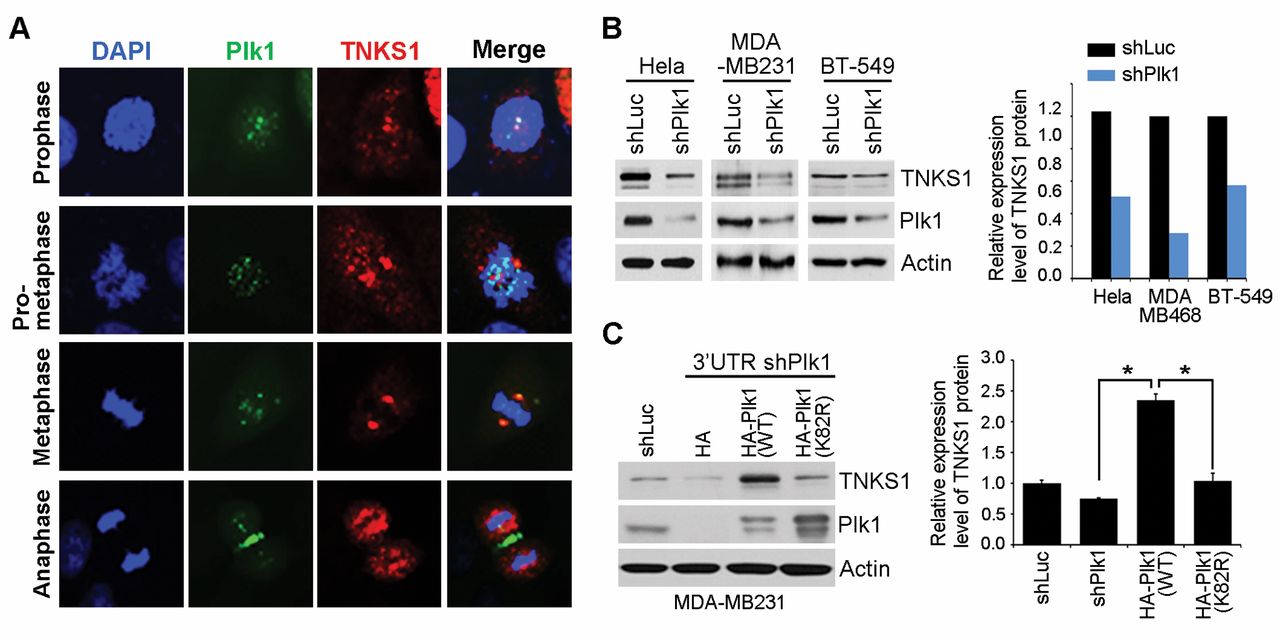
Plk1 regulates TNKS1 protein stability in breast cancer cells. (A) Immunofluorescence with anti- tankyrase 1 and anti-Plk1 antibody in MDA-MB231 cells. (B) Hela, MBA-MB-231, and BT-549 cells were transfected with shRNA specifically targeting luciferase (shLuc) or Plk1 (shPlk1). Forty-eight hours after transfection, and then the cells were harvested and Lysates were prepared and analyzed by immunoblotting using the indicated antibodies (left). TNKS1 expression level by plk1 depletion was quantified through scanning densitometry with actin as an internal control (right). (C) MDA-MB231 cells were co-transfected with shRNA specific for Plk1 3’-UTR (shPlk1 #2) and expression plasmids encoding shRNA-insensitive, HA-Plk1 WT or HA-Plk1 K82R (a kinase-dead mutant of Plk1). Thirty-six hours after transfection, lysates were prepared and subjected to immunoblot analysis using the indicated antibodies. *p<0.05 vs. indicated.
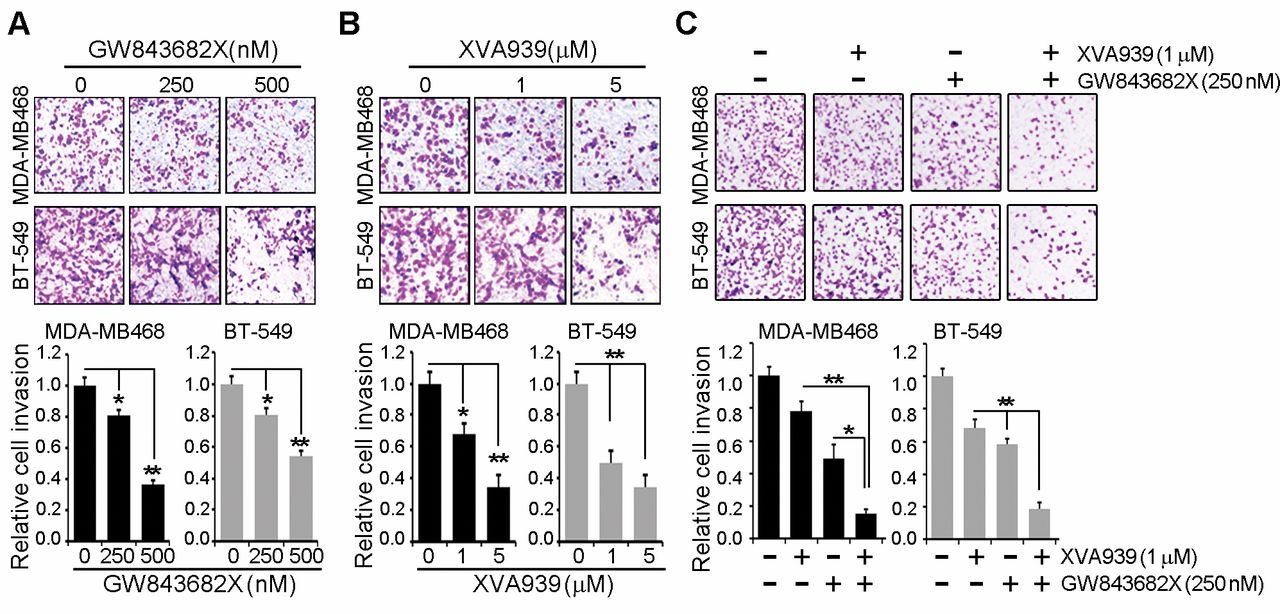
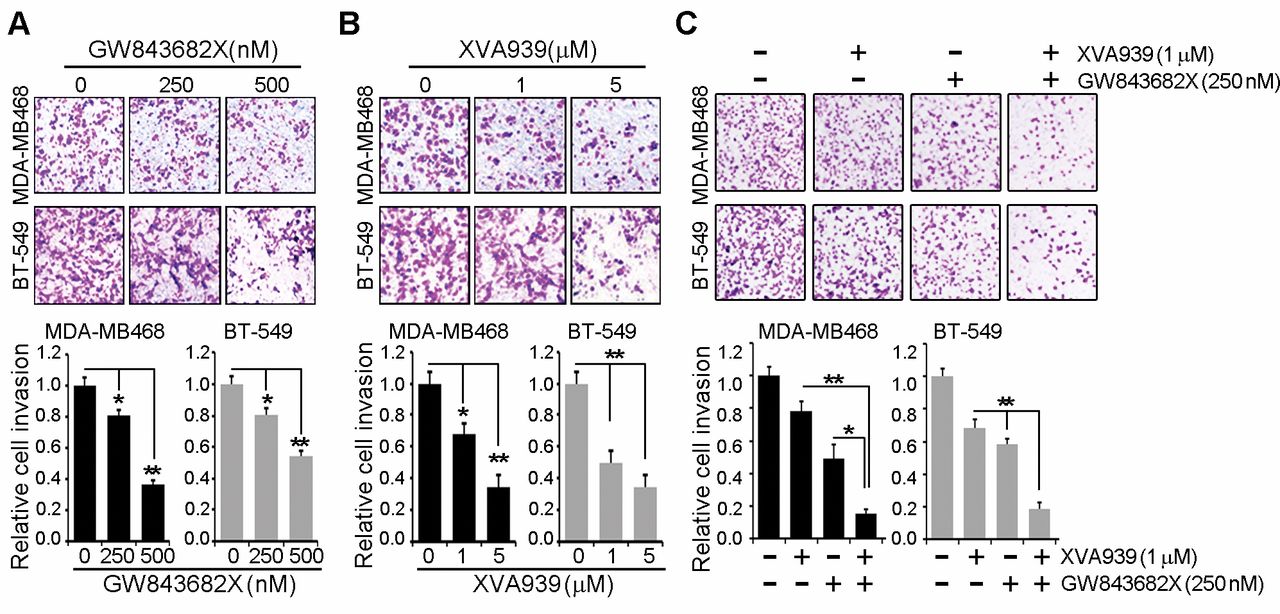
Inhibition both Plk1 and TNKS1 synergistically reduced the cell invasion in MDA-MB468 and BT-549 cells. (A) MDA-MB468 and BT-549 cells with GW843682X dose-dependent treatment (0, 250, 500 nM) and then were subjected to transwell invasion assays. (B) MDA-MB468 and BT-549 cells with XAV939 dose-dependent treatment and then were subjected to transwell invasion assays. (C) MDA-MB468 and BT-549 cells with 250 nM GW843682X and 1 μM XAV939 and then were subjected to transwell invasion assays. All data are shown as the means±S.D. of at least three independent experiments. The p-values were calculated using the unpaired Student's t-test. *p<0.05; **p<0.01 vs. indicated.

Inhibition both Plk1 and TNKS1 synergistically reduced the cell migration in MDA-MB468 and BT-549 cells. (A) MDA-MB468 and BT-549 cells with GW843682X dose-dependent treatment (0, 250, 500 nM) and then were subjected to transwell migration assays. (B) MDA-MB468 and BT-549 cells with XAV939 dose-dependent treatment (0, 1, 5 μM) and then were subjected to transwell migration assays. (C) MDA-MB468 and BT-549 cells with 250 nM GW843682X and 1 μM XAV939 and then were subjected to transwell migration assays. All data are shown as the means±S.D. of at least three independent experiments. *p<0.05; **p<0.01 vs. indicated.
PLK1 regulates TNKS1 protein stability. In a previous study, we demonstrated that inhibition or depletion of PLK1 decreased the stability and poly-ADP-ribosylation activity of TNKS1 in HeLa cells (15). Here, we investigated whether PLK1 co-localized with TNKS1 in MDA-MB231 cells. As shown in Figure 2A, TNKS1 co-localized with PLK1 at spindle poles and the spindle midzone. Next, we tested whether depletion of PLK1 affected the stability of TNKS1 in breast cancer cell lines. Interestingly, TNKS1 protein stability was reduced when PLK1 expression was specifically inhibited in HeLa, MBA-MB-231, and BT-549 cells (Figure 2B). We depleted endogenous PLK1 in MDA-MB231 cells, and then, overexpressed shRNA-insensitive HA-tagged PLK1 wild-type (WT) or kinase-dead PLK1 K82R mutants. Overexpression of PLK1 WT significantly increased the protein levels of TNKS1, compared to that in cells overexpressing PLK1 K82R (Figure 2C). Together, these results indicated that depletion of PLK1 decreased the stability of TNKS1 protein in breast cancer cell lines.
Effect of inhibition of TNKS1 or PLK1 on breast cancer cell invasion and migration. Previous studies have demonstrated a role of PLK1 in the migration and invasion of colorectal, lung, and breast cancer cells (6). To investigate the effect of PLK1 inhibition on migration and invasion, MDA-MB468 and BT-549 cells were treated with PLK1 inhibitor, GW843682X. As shown in Figure 3A, inhibition of PLK1 significantly reduced cell invasion, compared to that in control cells treated with DMSO. Next, we tested the effect of TNKS1 inhibition by XAV939 treatment on the migration and invasion capacities of MDA-MB468 and BT-549 cells. Similar to PLK1 inhibition, inhibition of TNKS1 dramatically decreased invasion in cells treated with XAV939 (Figure 3B). Moreover, combination treatment of cells with the TNKS1 inhibitor and PLK1 inhibitor potently and synergistically reduced cell invasion (Figure 3C). Similar results were observed in the migration assays (Figure 4). Together, these results indicated that inhibition of TNKS1 or PLK1 dramatically reduced the cell invasion and migration of breast cancer cells.
Pharmacological targeting of PLK1 and TNKS1 synergistically decreases cell survival in TNBC. We observed that the effect of PLK1 inhibitor treatment on cell viability varied across different cancer cell lines. As shown in Figure 5A, the viabilities of MDA-MB468 and BT-549 cells were significantly reduced, compared to that in three other breast cancer cell lines, MCF10A, MCF7, and SK-BR-3. However, the viability of MDA-MB231 cells was not affected by PLK1 inhibitor treatment, compared to that in the same breast cancer cell lines. Previous studies have indicated that the level and activity of β-catenin contribute to the sensitivity of breast cancer cells to chemotherapeutic agents (17-19). Thus, we analyzed β-catenin expression in the different breast cancer cell lines. Interestingly, MDA-MB231 cells expressed lower levels of β-catenin than MDA-MB-468 or BT-549 cells (Figure 5B). Previous evidence reports that combination inhibition of PLK1 and TNKS1 significantly increases apoptotic response in prostate cancer, in comparison to responses elicited from PLK1 or TNKS1 inhibition alone (20). Therefore, we investigated the effect of combination treatment with TNKS1 inhibitor and PLK1 inhibitor on cell viability. As shown in Figure 5C, combination treatment synergistically inhibited cell growth in MDA-MB-231 cells, compared to that in cells treated with TNKS1 inhibitor or PLK1 inhibitor alone. Similar results were observed in the migration and invasion assays (Figure 5G). Furthermore, as demonstrated by Annexin V/PI staining (Figure 5D), TUNEL assay (Figure 5E), and western blot analysis (Figure 5F), this combination therapy consistently induced higher apoptotic levels than that observed when either inhibitory agent was used alone in MDA-MB231 cells. Taken together, these results indicated that the combined inhibition of PLK1 and TNKS1 led to enhanced anticancer effects, suggesting that it may improve the chemotherapeutic treatment of TNBC.
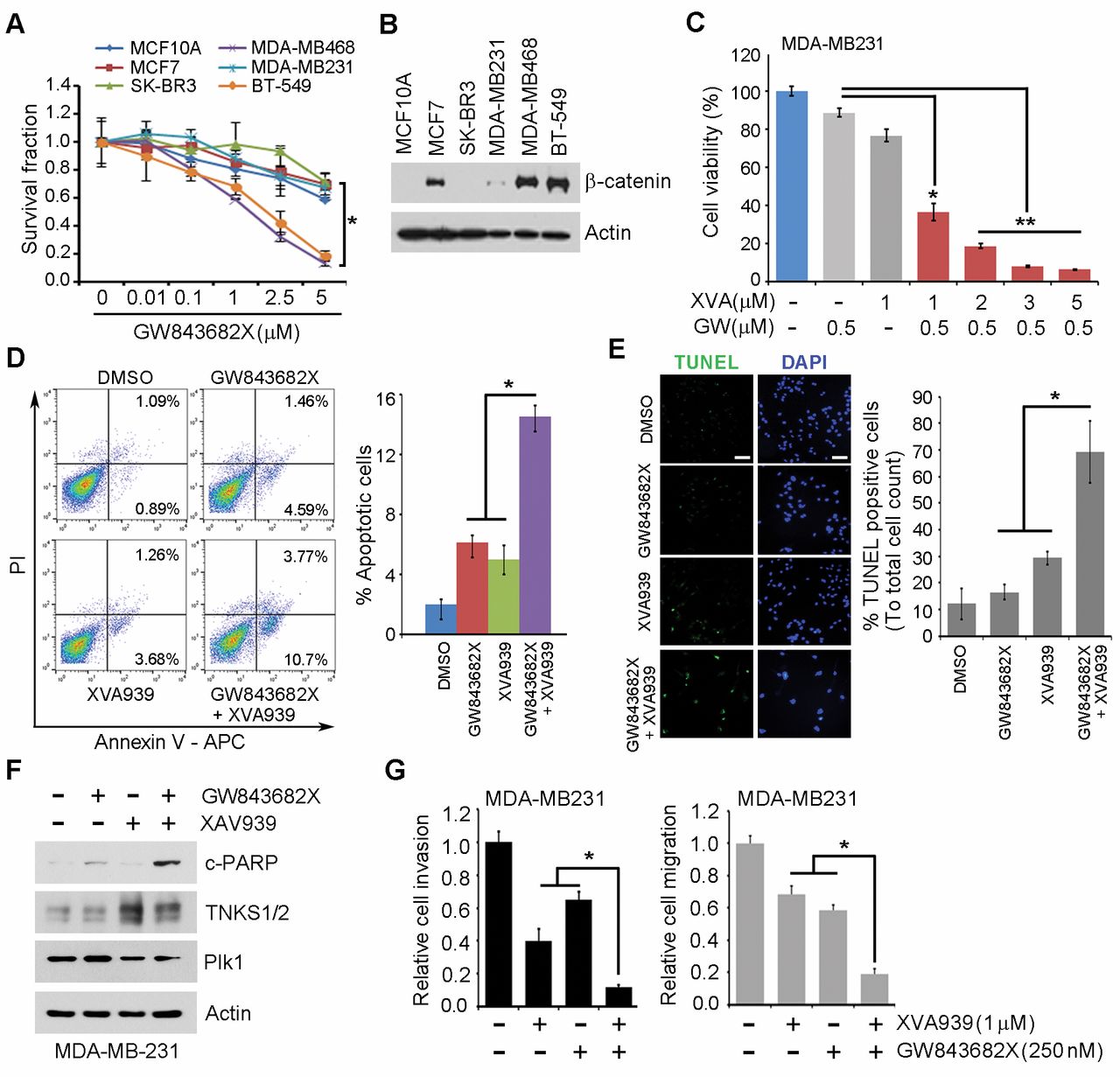
Combination inhibition of TNKS1 and Plk1 resulted in induced chemosensitization in triple-negative breast cancer (TNBC) cells. (A) Six different breast cancer cell lines with GW843682X dose-dependent treatment. At 7 days after treatments, cells were incubated with crystal violet coloration, and the intensity of crystal violet was determined. The percentage of surviving fractions was determined by the intensity of crystal violet relative to DMSO. (B) Six different breast Cancer cells were analyzed for β-catenin expression using immunoblotting. (C) MDA-MB31 cells were treated with combination XAV939 and GW843682X. At 7 days after treatments, cells were incubated with crystal violet coloration, and the intensity of crystal violet was determined. MDA-MB31 cells were treated with combination XAV939 and GW843682X for 36h, and then analyzed the apoptosis by an Annexin-V/ PI (propidium iodide) apoptosis assay (D), TUNEL assay (E), and western blot analysis (F). (G) MDA-MB231 cells with 250 nM GW843682X and 1μM XAV939 and then were subjected to transwell invasion and migration assays. *p<0.05; **p<0.01 vs. indicated.
Discussion
Breast cancer is a highly heterogeneous disease owing to variations in genetic and histological features; breast cancers can be classified into six to ten subtypes (21). TNBC is a rare subtype of breast cancer, characterized by lack of expression of ER, PR, and HER2. Patients with TNBC are diagnosed with shorter overall survival than patients with other subtypes of breast cancer, and currently available therapies are ineffective and limited (22). Primary reasons for the failure of current TNBC treatment are the metastatic potential and intrinsic resistance to standard chemotherapies of TNBC (23). In addition, aberrant regulation of the Wnt/β-catenin signaling pathway in TNBC has been correlated to the tumorigenesis and metastasis of tumors, resulting in poor clinical outcome and secondary metastases to other regions of the body (11, 24). Therefore, novel therapeutic approaches that can effectively target TNBC need to be developed to combat drug resistance and breast cancer recurrence by metastasis. In this study, we evaluated the therapeutic effects of combination inhibition of PLK1 and TNKS1, and we present the potential benefits of employing this as a new therapeutic strategy for the treatment of TNBC.
PLK1 is overexpressed in various human cancers, including breast cancer, and PLK1 overexpression has been related to tumor chemoresistance and poor prognosis of cancer patients (6, 25). In tumor development, silencing of PLK1 activity induces mitotic cell cycle arrest and enhances drug sensitivity and, consequently, apoptotic cell death in several cancers (8, 26). In a previous study, we demonstrated that PLK1 bound to and phosphorylated TNKS1. Phosphorylation of TNKS1 by PLK1 was shown to be important for TNKS1 protein stability and telomeric poly (ADP-ribose) polymerase (PARP) activity (15). Recently, many studies have reported that TNKS1 is upregulated in several cancers, and cancer therapy using TNKS1 inhibitors is one of the most promising options for cancer therapeutics targeting Wnt/β-catenin and telomere-based signaling (27, 28). Thus, we conducted combined chemotherapeutic treatment of PLK1 and TNKS1 inhibitors and evaluated their effects on cell viability and metastasis in TNBC. We observed that PLK1 was highly expressed in TNBC cell lines and depletion of PLK1 reduced TNKS1 protein levels, whereas overexpression of PLK1 enhanced TNKS1 protein levels. In contrast, the kinase-dead PLK1 K82R mutant did not increase TNKS1 protein expression. In comparison to treatment with each inhibitor alone, combined inhibition of PLK1 and TNKS1 at lower doses synergistically attenuated TNBC cell proliferation, migration, and invasion. Our data demonstrated that the kinase activity of PLK1 may be important for TNBC metastasis and TNKS1 protein stability; therefore, combination therapy targeting both PLK1 and TNKS1 may be effective in the treatment of metastatic breast cancer.
Chemotherapy using PARP or HDAC inhibitors is emerging as a novel therapeutic option to treat metastatic breast cancers, such as TNBC and tumors with BRCA mutations, by blocking DNA repair pathways while increasing the DNA damage burden of cells (29, 30, 31). Over half of patients with BRCA-mutated, metastatic breast cancer have tumors classified as triple-negative, and clinical trials to assess the efficacy and safety of a TNKS inhibitor in metastatic TNBC patients are currently being conducted (32, 33). TNKS1 was first identified as a TRF-binding protein and a member of the PARP family (34). Numerous studies have shown that inhibition of TNKS1 attenuates Wnt/β-catenin signaling through the stabilization of Axin in breast cancer cells (27). Interestingly, although inhibition of PLK1 did not affect cell viability in TNBC lines that expressed low levels of β-catenin, combination treatment with PLK1 inhibitor and a low dose of TNKS1 inhibitor dramatically reduced cell viability (Figure 5A). Furthermore, treatment with PLK1 inhibitor in combination with TNKS1 inhibitor significantly augmented cell apoptosis and PARP cleavage in TNBC cells that expressed low levels of β-catenin (Figure 5B-F). These results suggested that combination therapy of PLK1 and TNKS1 inhibitors may be a potential therapeutic strategy for the treatment of TNBC and other breast cancers, independent of β-catenin expression.
In conclusion, our results indicated that co-inhibition of PLK1 and TNKS1 led to chemosensitization of TNBC cells. Our observations of this novel combination therapy provide new insights into improving current therapeutic strategies to target PLK1-resistant TNBC in the future.
Acknowledgements
This work was supported by the National Research Foundation of Korea(NRF) Grant funded by the Korean Government(MSIP) (NRF-2015R1D1A1A01058623, 2016R1A5A2945889 and 2016R1 6A3A11930537).
Footnotes
This article is freely accessible online.
Conflicts of Interest
All Authors have no conflicts of interest to disclose.
- Received December 20, 2017.
- Revision received January 27, 2018.
- Accepted January 29, 2018.
- Copyright© 2018, International Institute of Anticancer Research (Dr. George J. Delinasios), All rights reserved





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}