


Abstract
Aim: Naftopidil is used to treat benign prostate hyperplasia. Moreover, previous studies have shown that naftopidil reduced viability of many types of cancer cells. Therefore, we investigated the antitumor mechanism of naftopidil in this study. Materials and Methods: We used the HGC27 human gastric cancer cell line. It was treated with naftopidil, pan-caspase inhibitor, and chloroquine diphosphate (CQ). Cell viability and cell death were investigated by the assay and annexin V/ propidium iodide assay. Phosphorylation of protein kinase B (AKT) (Ser473) was measured by western blotting. Alteration of light chain 3B (LC3B) was investigated by western blotting and immunofluorescence. Results: Naftopidil reduced phospho-AKT (Ser473) and altered LC3B. Combination of naftopidil and CQ reduced cell viability and phospho-AKT (Ser 473). Conclusion: Naftopidil induces apoptosis and autophagy of HGC27 cells, however, autophagy is considered to inhibit apoptosis. We concluded naftopidil and CQ have a synergistic antitumor effect.
Alpha-1 adrenoceptor (AR) blockers are widely used as therapeutic agents for dysuria. Recent retrospective observational cohort studies showed that AR blockers, such as doxazosin and terazosin, reduced the risk of bladder and prostate cancer (1, 2). Therefore, AR blockers are thought to be probable drug repositioning candidates in urological cancer therapy. Naftopidil is an AR blocker that reduce symptoms of urination disorder caused by benign prostatic hyperplasia (3). Like other AR blockers, naftopidil reduces the risk of prostate cancer (4). Moreover, naftopidil was found to induce death of prostate cancer cells (5, 6). We previously reported that naftopidil-induced cell death was observed not only in prostate cancer cells but also in bladder and renal cancer cells in vitro (7). Furthermore, our in vivo study showed that the antitumor effect of naftopidil had high selectivity for bladder cancer cells (8). Therefore, we consider that naftopidil is a prospective therapeutic agent against bladder cancer. On the other hand, our previous study showed that there are no relationships between inhibition of cell viability and AR blocking by naftopidil (8), while the mechanism of cell death induced by naftopidil is not clear. We hypothesized that apoptosis and autophagy were essential for the antitumor effect of naftopidil.
Apoptosis is a form of typical programmed cell death. Phosphatidylinositol 3-kinase (PIK3)–serine/threonine protein kinase B (PKB/AKT) signaling pathway is involved in inducing apoptosis (9). PKB/AKT is a key regulator of a wide range of cellular processes including growth, proliferation and survival (10). Activation of AKT is dependent upon signaling by heterodimeric phosphatidylinositol 3-kinase (PI3K) (11). Some studies demonstrated that autophagy could induce or inhibit cell death in gastric cancer cells (12, 13). Autophagy is a survival-promoting pathway that captures, degrades, and recycles intracellular proteins and organelles in lysosomes (14).
In this study, we investigated whether naftopidil induces apoptosis and autophagy in HGC27 cells, a human gastric cancer cell line which was found to be the most sensitive to naftopidil out of various types of cancer cells that we tested (Table I).
Materials and Methods
Materials and reagents. Fetal bovine serum (FBS) was purchased from Biowest (Nuaillé, France). 3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) and Hoechst33342 were purchased from Dojindo Laboratories (Kumamoto, Japan). Annexin V-FITC (fluorescein isothiocyanate) Kit was purchased from MBL (Aichi, Japan). Propidium iodide (PI) was purchased from Sigma Aldrich (Darmstadt, Germany). Pan-caspase inhibitor benzyloxycarbonylvalyl-alanyl-aspartyl fluoromethyl ketone (z-VAD-FMK), was purchased from Enzo Life Sciences (Farmingdale, NY, USA). Naftopidil, and autophagy inhibitor chloroquine diphosphate (CQ) were purchased from Tokyo Kasei industry (Tokyo, Japan). Formalin neutral buffer was purchased from Kenei-Pharm (Osaka, Japan). RPMI-1640 medium and penicillin-streptomycin mix solution were purchased from Life Technologies Japan (Tokyo, Japan). N-Dimethylformamide (DMF), sodium dodecyl sulfate (SDS) and Triton X-100 were purchased from Nacalai Tesque (Kyoto, Japan). Phosphate-buffered saline (PBS) was purchased from Santa Cruz Biotechnology (Dallas, TX, USA). EzRun and EzFastBlot were purchased from ATTO (Osaka, Japan) and used as SDS-PAGE buffer and semi dry blotting buffer. EzTBS was purchased from ATTO (Osaka, Japan). Bovine serum albumin (BSA), Low Salt and Formalin Neutral Buffer Solution were purchased from Wako (Osaka, Japan). DyLight 488-conjugated anti-rabbit IgG antibody was purchased from Rockland Immunochemicals (Limerick, PA, USA). Alexa 594-conjugated anti-rabbit IgG antibody was purchased from Molecular Probes (Eugene, OR, USA).
Effect of naftopidil on individual cell lines. These cells were treated with 0-50 μM naftopidil for 48 h. This table shows the 50% inhibitory concentration (IC50) for each cell line.
Cell culture. A human gastric cancer cell line (HGC27) (15), a human prostate cancer cell line (PC3), a human bladder cancer cell line (T24), a human renal cancer cell line (786O) were used in this study. HGC27 was obtained from RIKEN Cell Bank (Tsukuba, Ibaraki, Japan). PC3, T24, and 786O were obtained from the American Type Culture Collection (Manassas, VA, USA). They were cultured in RPMI-1640 medium supplemented with 10% (v/v) heat-inactivated FBS and 1.0% penicillin-streptomycin (final concentration: 100 U/ml, 100 μg/ml). Cultures were maintained with a humidified atmosphere of 5% CO2 and 95% air at 37°C in a CO2 incubator (Sanyo, Osaka, Japan).
Assay of cell viability. PC3, T24, 786O and HGC27 cells were seeded in 96-well plates (3000 cells/well) and cultured in RPMI-1640 medium with 10% FBS. On the following day, the medium was replaced with RPMI-1640 medium with 5% FBS and 50 μM (PC3, T24, 786O and HGC27 cells) or 1, 3, 10, 30, 100 μM (HGC27 cells) naftopidil, and plates were returned to the CO2 incubator for 48 hours (PC3, T24, 786O and HGC27 cells) or 2-48 h (HGC27 cells). Cell viability was assayed by MTT assay with SPECTRA max PLUS384 (Molecular Devices, Sunnyvale, CA, USA) as described previously (16). The percentage cell viability was calculated from the following absorbance measurements: An: negative control (no cells: 0%) cells; Ac: positive control (non-treated cells: 100% viability) cells, and Ax: drug-treated cells, as: (Ax−An)/(Ac−An) ×100.
Annexin V (AV)/PI assay. The cells were seeded in 6-well plates and treated with naftopidil or naftopidil and zVAD-FMK for 24 hours then harvested by trypsinization, washed once with cold PBS, and resuspended in 500 μl binding buffer. Subsequently, 10 μl AV and 5 μl PI were added to the cells. The cells were mixed and incubated for 15 min at room temperature in the dark and then analyzed by flow cytometry with an LSRFortessa X-20 instrument (Becton Dickinson and Company, Franklin Lakes, NJ, USA). The same experiment was repeated three times.
Western blotting analysis. HGC27 cells were lysed in SDS buffer at room temperature and sonicated by Digital Sonifier (BRANSON, Danbury, CT, USA). The samples were stored at −80°C until analyzed. The samples were loaded on SDS–PAGE gel and transferred to polyvinylidene difluoride membrane. After blocking with TBST containing 5% (w/v) of BSA, the membrane was reacted with antibodies (all diluted 1:5000, from Cell Signaling Technology, Boston, MA, USA) against p-AKT Ser473, p-AKT substrates, or light chain 3B (LC3B), followed by a conjugated anti-rabbit IgG antibody. For glyceraldehyde-3-phosphate dehydrogenase (GAPDH) detection, blotting membrane was reacted with HRP-conjugated anti-GAPDH (1:20000; Medical & Biological Laboratories, Aichi, Japan).
Indirect immunofluorescence. HGC27 cells cultured in 6-well plates were fixed for 15 min at room temperature with 10% Formalin Neutral Buffer Solution (Wako) and blocked with 0.3% Triton X-100 (Nacalai Tesque, Kyoto, Japan), 5.0% BSA (Wako) in PBS for 120 minutes at room temperature. The cells were incubated with anti-LC3B (1:400; Cell Signaling Technology) in 0.3% Triton X-100 and 1.0% BSA containing PBS overnight at 4°C. Then the cells were incubated for 60 minutes at room temperature with DyLight 488-conjugated anti-rabbit IgG antibody (1:500; Rockland Immunochemicals), Alexa 594-conjugated anti-rabbit IgG antibody (1:5000; Molecular Probes), and Hoechst33342 (1:2,000; Dojindo, Kumamoto, Japan). The cells were washed three times with PBS. Images were taken with a Zeiss LSM 780 inverted confocal laser scanning microscopy with a 20× objective lens and processed by ZEN 2.3 image processing software (Zeiss, Oberkochen, Germany).
Statistical analysis. All data are expressed as the mean±standard error of the mean (SEM). Statistical comparisons between groups were performed using Student's t-test (indicated in each figure legend), where values of p<0.05 were considered statistically significant.
Results
Naftopidil induces apoptosis. In a previous report, we showed that naftopidil had an antitumor effect and reduced viability of cancer cells (8). In this study, we tested the effects of naftopidil on viability of PC3, T24, 786O and HGC27 cells by MTT assay. Table I shows that HGC27 cells were the most sensitive to naftopidil among these cell lines, therefore we decided to use HGC27 cells in this study. Next, we tested the effect of naftopidil on viability of HGC27 cells. Regardless of the concentration, no decrease in cell viability by naftopidil treatment was observed within 16 h. We calculated that the 50% inhibitory concentration (IC50) value for naftopidil against HGC27 cells was 62 μM for 24 hours and 17.9 μM for 48 h (Figure 1A).
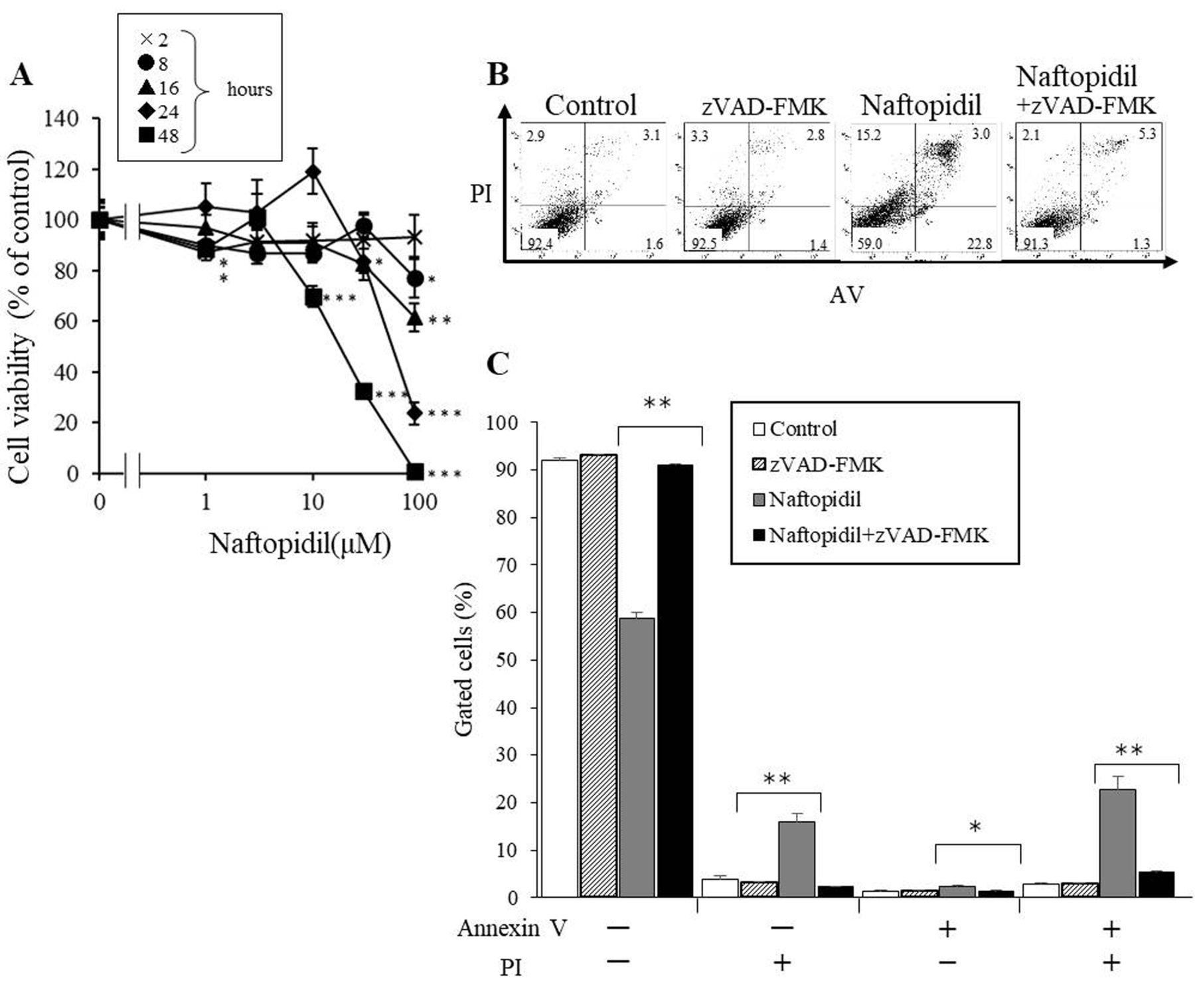
Effect of naftopidil on cells. A: HGC27 cells were treated with different concentrations of naftopidil for 2-48 h. Significant difference in cell viability between groups treated with naftopidil or not at *p<0.05, **p<0.01, and ***p<0.001. B and C: HGC27 cells were treated with 50 μM of pan-caspase inhibitor zVAD-FMK, 50 μM naftopidil, or 50 μM zVAD-FMK and 50 μM naftopidil for 24 h. After staining with annexin V (AV)/propidium iodide (PI) flow cytometric analysis was performed. Significantly different at *p<0.05 and **p<0.01. The data shown represent the mean±SEM.
Evading apoptosis is a major means by which cancer cells are protected from cell death. In other words, induction of apoptosis is an important way to kill cancer cells. In this study, we further tested whether naftopidil induced apoptosis in HGC27 cells with AV/PI assay. The results show the rates of living AV-negative and PI-negative), necrotic (AV-negative and PI-positive), early apoptotic (AV-positive and PI-negative), and late apoptotic (AV-positive and PI-positive) cells. After HGC27 cells were treated with 50 μM naftopidil alone for 24 h, both early and late apoptotic rates were increased (Figure 1B and C), while after treatment with 50 μM of pan-caspase inhibitor zVAD-FMK and 50 μM naftopidil, the rate or late apoptosis of HGC27 cells were reduced more than after treatment with 50 μM naftopidil alone.
Naftopidil reduces AKT expression and AKT substrates activation. When AKT is phosphorylated and activated, it inhibits apoptosis. We tested phospho-AKT (Ser 473) to reflect the degree of apoptosis. Phospho-AKT (Ser 473) in HGC27 cells was decreased by naftopidil treatment (Figure 2A and B). When HGC27 cells were treated with 30 μM of naftopidil, phospho-AKT in HGC27 cells showed time dependency (Figure 2A). Moreover, dose-dependent decrease of AKT phosphorylation was also observed with 24 h of naftopidil treatment (Figure 2B). Naftopidil also reduced phospho-AKT substrates (Figure 2C). These results showed that naftopidil inactivated both AKT and its substrates.

Effect of naftopidil on protein kinase B (AKT) and its substrates in HGC27 cells. The data shown represent the mean±SEM. HGC27 cells were treated with 30 μM naftopidil at the indicated times (A) or with the indicated concentration for 24 hours (B and C). The levels of p-AKT (B) and p-AKT substrate (C) were detected by immunoblotting. *Significant difference in cell viability between control and naftopidil-treated cells at p<0.05.
Naftopidil appears to induce autophagy. We tested whether naftopidil induced autophagy in HGC27 cells via observation of LC3-II. After HGC27 cells were treated with naftopidil at 10-30 μM for 24 hours, the protein level of LC3-II was increased, while the level of LC3-I decreased in a naftopidil dose-dependent manner (Figure 3A). We also examined the intracellular localization of LC3B in HGC27 after 24 hours of naftopidil treatment, and many LC3B-positive dots were observed in cytoplasm (Figure 3B).
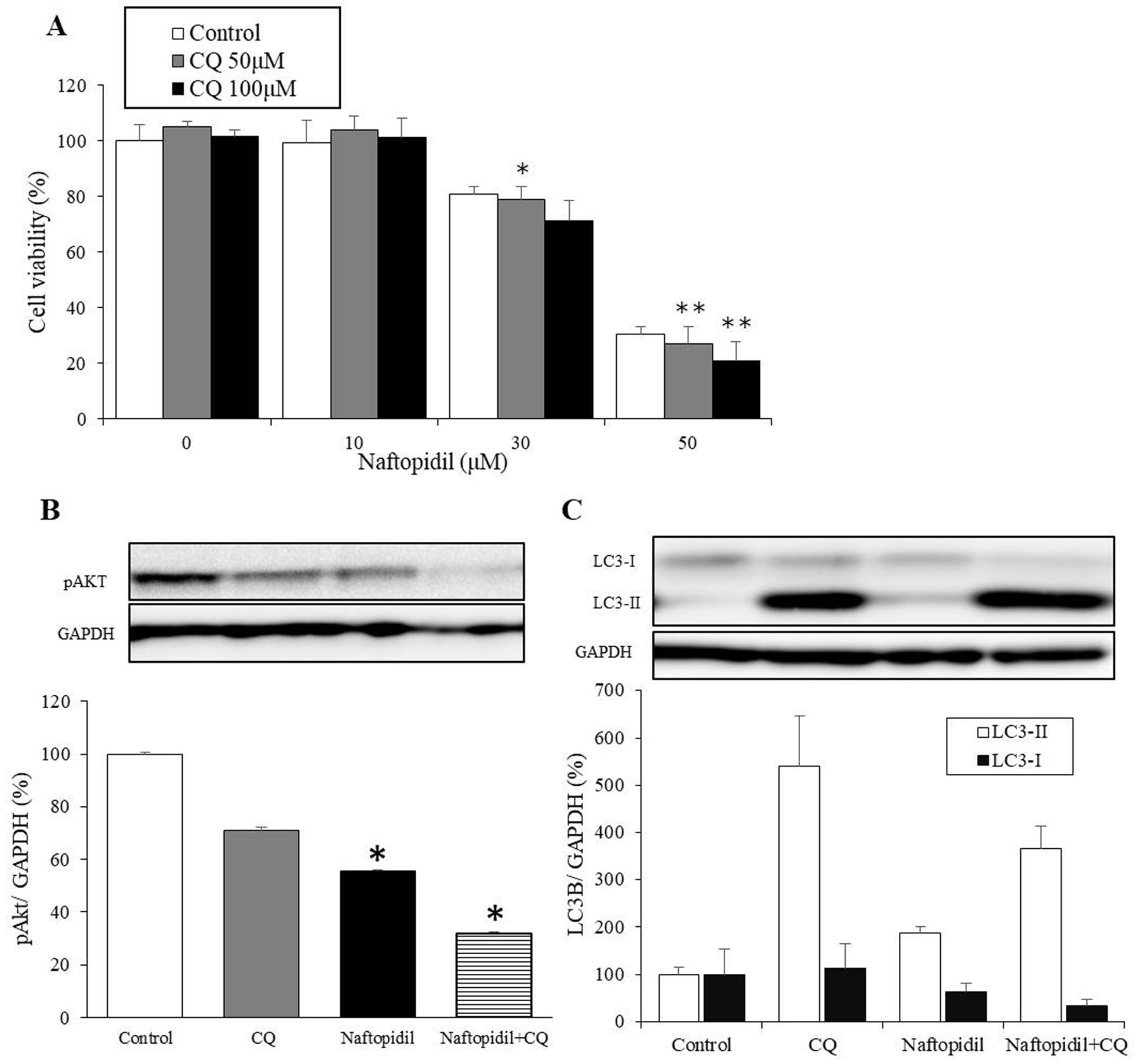
Inhibition of autophagy promotes naftopidil-induced apoptosis. We tested whether naftopidil-induced apoptosis and autophagy were related or not by MTT assay. When cells were treated with naftopidil and autophagy inhibitor CQ for 24 hours, we found that cell viability deceased more than with naftopidil alone (Figure 4A). Furthermore, cell viability at 24 h after treatment with 30 μM naftopidil and 50 μM CQ was significantly lower than after treatment with 30 μM of naftopidil alone. Similarly, 50 μM naftopidil with 50 μM CQ or 100 μM CQ significantly reduced viability compared with treatment with 50 μM of naftopidil alone (p<0.05 and p<0.01, respectively) (Figure 4A). After cells were treated with 30 μM of naftopidil and 50 μM of CQ for 24 h, we found that phospho-AKT (Ser473) significantly decreased more than after treatment with 30 μM of naftopidil alone (p<0.025) (Figure 4B). We also noted that naftopidil increased the level of LC3-II and reduced that of LC3-I, and naftopidil with CQ also increased the level of LC3-II dramatically and reduced that of LC3-I (Figure 4C).

Effect of naftopidil on light chain 3B (LC3B) in HGC27 cells. The data shown represent the mean±SEM. HGC27 cells were treated with different concentration of 10-30 μM naftopidil for 24 h. The levels of LC3B (LC3-I and LC3-II) were detected by immunoblotting (A), and the localization of LC3B was detected by immunofluorescence (B). *Significant difference in cell viability between control cells and those treated with 30 μM naftopidil at p<0.05.
Discussion
In this study, we investigated the mechanism of anticancer effect of naftopidil. z-VAD-FMK reduced the proportion of cells that were apoptotic when treated with naftopidil (Figure 1B). This shows that naftopidil reduces cell viability by inducing apoptosis. Decrease of phospho-AKT (Ser473) was observed in naftopidil-treated HGC27 cells. Phosphorylated AKT is necessary for the activation of AKT substrates (17). In other words, naftopidil-induced decrease of AKT phosphorylation appears to be involved in apoptosis induction via inhibition of the PI3K–AKT pathway. Our results suggest that naftopidil induced apoptosis of HGC27 cells. Many types of AR blockers such as naftopidil, prazosin and doxazosin reduce in cancer cell viability (8), particularly naftopidil, which led to the most significant decrease of cell viability in cancer cells and the lowest toxicity against normal cells (8). Another study of ours showed that naftopidil reduced cell viability regardless of AR blocking in human malignant mesothelioma cells (18), and induced apoptosis through G1 arrest in human bladder cancer cells (18). We previously observed that naftopidil induced sub G1 arrest (data not shown). From these results, we concluded that the antitumor effect of naftopidil is based on apoptosis induction in cancer cells. Naftopidil reduced phospho-AKT (Ser 473) after 24 hours, however, it hardly decreased within 12 h. This might mean that naftopidil influences upstream pathway from AKT signaling until 12 hours.

Relationship of apoptosis and autophagy. A: HGC27 cells were treated with different concentrations of naftopidil alone or naftopidil with autophagy inhibitor chloroquine diphosphate (CQ) for 24 h. The data shown represent the mean±SEM. Significant difference in cell viability between naftopidil alone and naftopidil with CQ at *p<0.05 and **p<0.01. B and C: HGC27 cells were treated with 50 μM of CQ, 30 μM of naftopidil, or 50 μM naftopidil and 30 μM CQ for 24 hours. The levels of p-AKT (B) and LC3-II and LC3-I (C) were detected by immunoblotting. *Significant difference in cell viability between cells treated with naftopidil alone and those treated with naftopidil with CQ at p<0.05.
Naftopidil treatment caused decrease of LC3-I and increase of LC3-II. These results suggest that naftopidil induced the transformation from LC3-I to LC3-II (Figure 3A). Moreover, results of immunofluorescence showed that naftopidil treatment increased the frequency of LC3B-positive dots in cytoplasm, and we consider that these LC3B-positive dots were autophagosome (Figure 3B). Therefore, our present results suggest that naftopidil induces autophagy through increase of LC3-II and the shift from LC3B to autophagosome.
CQ is a suppressor of autolysosome formation. We found that the combination of naftopidil and CQ reduced the phospho-AKT (Ser473) more than naftopidil alone, and treatment of CQ alone increased expression of LC3-II dramatically, nevertheless, it did not reduce that of LC3-I (Figure 4C), whereas, naftopidil alone increased expression of LC3-II and reduced that of LC3-I. Taken together, we considered that autolysosome is conducted and LC3-II is resolved in naftopidil-treatment cells. These results show that autophagy via treatment with naftopidil did not promote but in fact reduced cell viability by inhibiting apoptosis of HGC27 cells, and indicate that when cancer cells are treated with naftopidil and an autophagy inhibitor such as CQ, there may be a synergistic antitumor effect.
In conclusion, naftopidil has an antitumor effect that is caused by apoptosis induction, and the antitumor effect of naftopidil appears to be stronger when it is used with autophagy inhibitor.
Acknowledgements
The Authors wish to thank Satoko Kodama for her help in the completion of the article.
Footnotes
Conflicts of Interest
The Authors have no potential conflicts of interest to disclose in regard to this study.
- Received November 6, 2017.
- Revision received December 7, 2017.
- Accepted December 11, 2017.
- Copyright© 2018, International Institute of Anticancer Research (Dr. George J. Delinasios), All rights reserved
References
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.