


Abstract
Ultraviolet (UV)-induced DNA lesions are almost exclusively removed by the nucleotide excision repair (NER) pathway, which is essential for prevention of skin cancer development. Patients with xeroderma pigmentosum (XP) are extremely sun sensitive due to a genetic defect in components of the NER cascade. They present with first signs of premature skin aging at an early age, with a considerably increased risk of developing UV-induced skin cancer. XP belongs to the group of DNA repair defective disorders that are mainly diagnosed in the clinic and in hindsight confirmed at the molecular level. Unfortunately, there are no causative treatment options for this rare, autosomal-recessive disorder, emphasizing the importance of an early diagnosis. Subsequently, UV-protective measures such as the reduction of exposure to environmental UV and regular skin cancer screenings should be undertaken to substantially improve prognosis as well as the disease course.
- Xeroderma pigmentosum
- nucleotide excision repair
- interdisciplinary treatment
- clinical and molecular diagnostics
- review
The human genome is constantly exposed to endo- and exogenous substances that threaten the integrity of DNA. The cell has several evolutionarily-conserved mechanisms to protect itself against damage to the genome (1). For this purpose, several repair mechanisms involving more than 130 DNA repair enzymes continuously monitor the DNA looking for damage (2). The nucleotide excision repair (NER) pathway is the only mechanism that detects and repairs damage leading to bulky distortions in the DNA backbone. Ultraviolet (UV) light is one of the most common exogenous causes of this sort of DNA damage (3). Patients with xeroderma pigmentosum (XP) have defective NER due to a gene defect in one of the components involved in this repair cascade (4).
Clinical Symptoms
Mutations in XP genes result in at least six different clinical entities: XP; XP plus neurological symptoms, trichothiodystrophy, XP plus trichothiodystrophy symptoms, XP plus Cockayne syndrome symptoms, and the cerebro-oculo-facial skeletal syndrome (5). Contrary to common assumptions, only about 60% of all patients with XP show increased sun sensitivity in the sense of severe dermatitis solaris after low UV exposure, which is usually the first conspicuous XP symptom during the first weeks of life. In 40% of patients with XP, there is no strong sun sensitivity. An early hyperpigmentation in sun-exposed areas, however, is common to all patients. In addition, signs of premature aging of the skin, and poikilodermic skin changes, can be observed as ealy as at the age of 3 to 5 years. Patients with XP develop basal cell and squamous epithelial cell carcinoma, as well as cutaneous melanoma, with an average of 8 years of age (6). In the general population, however, the first UV-induced malignancy develops much later, at about 60 years of age. In summary, patients with XP have a 10,000-fold increased risk for basal cell and squamous cell carcinomas and a 2,000-fold increased melanoma risk (7).
Furthermore, changes occur in UV-exposed areas of the eye (conjunctiva, cornea, lens and eyelids). In particular, conjunctivitis, cataract formation and pterygium formation, as well as rare tumors, are more frequently observed (Figure 1) (8).
In 25% of cases, patients with XP also develop neurological symptoms, which can vary greatly in their severity and time of first appearance. These include attenuated or missing tendon reflexes, progressive hearing loss, speech and gait disturbances, as well as cognitive decline (9).
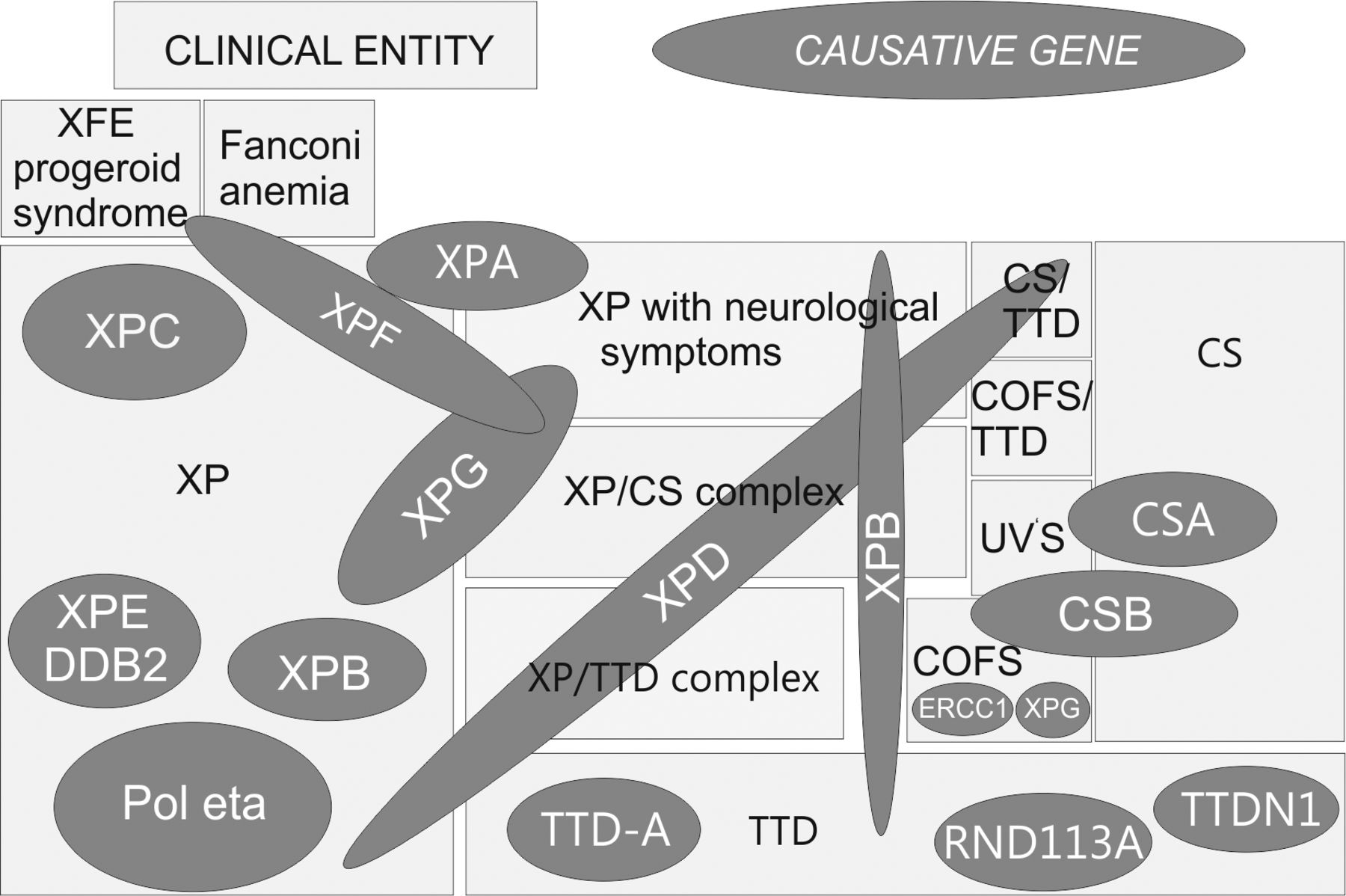
Twelve clinical entities and 14 molecular defects show the complex genotype–phenotype correlation of DNA-repair deficiency disorders. Different mutations in various genetic regions of DNA-repair proteins can lead to 12 different clinical entities. Mutations in different genes can result in the same clinical entity. COFS: Cerebro-oculo-facial-skeletal syndrome, CS: Cockayne syndrome, TTD: trichothiodystrophy, UVS: UV-sensitive syndrome, XP: xeroderma pigmentosum. Illustration by Dr. rer. nat. Christina Seebode, modified from (5).
Disease-causing Genes and Epidemiology
To date seven different XP complementation groups XP-A to XP-G, corresponding to the underlying gene defect (mutations in the genes XPA-XPG), plus a variant form in just under 10% of all patients with XP with a mutation in the translational polymerase η gene (PolH), have been detected (4). These complementation groups occur at different frequencies and differ in the severity of the symptoms in regard to skin cancer development and neurological involvement (see Table I). The disease is characterized by pleiotropy, meaning that different mutations in a gene result in the manifestation of several different phenotypic features. Due to the involvement of the XP proteins in various essential cellular processes, such as DNA repair and transcription, genotype–phenotype correlations are particularly complicated (see Figure 1). XP is an autosomal-recessive disease and occurs worldwide in all ethnic groups and skin types (10). The incidence in the US and Europe is one in one million. In geographically isolated populations such as in North Africa or Japan, the incidence is 10-fold higher (11, 12).
Diagnostics
DNA-repair defect syndromes are primarily diagnosed in the clinic and confirmed by functional cell-based test systems, as well as genetic sequencing. The diagnosis should be made as early as possible in order to be able to take separate UV-protective measures at an early stage. The diagnosis of patients with XP requires the cooperation of various clinical specialties such as dermatology, ophthalmology, ear, nose and throat medicine (otolaryngology), neurology, and imaging methods, as well as human genetics (see Figure 2) (13). In order to support this interdisciplinary patient care, our clinic is part of a European Reference Network for Rare Skin Diseases which was founded with the aim of connecting expert specialists of different rare skin syndromes to improve communication, diagnosis, and treatment guidelines. Thereby, patients all over Europe, also in the structurally weaker countries, can benefit from the expertise of all members. For the molecular diagnosis of XP, we apply functional DNA-repair assays, gene- and protein-expression analyses, as well as sequence analyses of the affected genes. The first step is the investigation of the post-UV survival of patient cells by means of a colorimetric assay for assessing cell metabolic activity [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay]. The patient cells can be obtained from blood (lymphocytes) or skin punch biopsies (fibroblasts) and are then irradiated with increasing UV doses. Without treatment, the cells grow normally but are killed by rising UV dose. Subsequently, cell survival is compared with cells from healthy donors and is usually clearly reduced in patients with XP.
Xeroderma pigmentosum (XP) complementation groups. Depicted in the following table is the frequency of the XP complementation groups, information on skin cancer number, neurological involvement, repair capability, the defective gene (XP genes A to G and polymerase eta) and chromosomal location. Adapted from (13).
In order to quantify the repair defect and functionally identify the mutated gene of the patient (without sequencing), the host cell reactivation (HCR) assay, a reporter gene assay or the so-called unscheduled DNA synthesis assay are used (4, 14). In the HCR test, the repair of a UVC-irradiated plasmid, and thus the NER capability, is investigated. The complementation groups can be functionally analyzed and determined by co-transfection of expression vectors in which the cDNA of XP genes is coded. The transfection of the XP cDNA, which is mutated in the patient, results in a significant increase in the repair capability of the patient's cells.
Once the affected protein has been identified, sequence analysis can reveal the exact nature of the mutation at the genomic level. All exons of a gene and neighboring intron regions are amplified and sequenced using the Sanger method. Genomic DNA required for this is obtained from blood or from fibroblasts of the patient. To confirm the diagnosis, further sequencing is performed at the cDNA level. Modern high-throughput methods (panel sequencing) can display mutations which, however, should be verified on the individual gene level as outlined above and must be distinguished from non-disease-triggering gene polymorphisms. Furthermore, the mRNA and protein expression levels can also be investigated.
In conclusion, the cells of patients with XP usually have a normal karyotype without large chromosomal aberrations. For affected families, it is also possible to carry out prenatal examinations from chorionic villi DNA, or amniotic fluid in the course of an amniocentesis. Here, knowledge of the disease-causing mutation is helpful sparing fetal DNA. This is normally the case as prenatal diagnostics is desired if an affected child was born in a previous pregnancy.
Therapy
At present, no causative therapy is available for patients with XP, which is why early diagnosis is of utmost importance. Immediately, strict and consistent sun protection and regular examination of the skin with treatment of pre-malignant lesions in the course of examinations for the early detection of skin cancer (quarterly) should be initiated.
Sun protection requires a broad avoidance of sun exposure, the use of sun protection (sun protection factor 50+), wearing long-sleeved clothing and wide-brimmed hats, as well as sunglasses and facial protection (15). For this purpose, special whole-body suits are available which block UV radiation to a certain extent. It is also advisable to use films with UV filters on window glass and to check the UV level in the environment and, thus, the daily exposure, with UV-measuring devices. Unfortunately to date, the neurological symptoms of the combined XP phenotypes can only be observed and supported during the disease course, an effective therapy is currently not available (16).
In the early detection of skin cancer, complete photo documentation of the entire skin surface is of particular importance in patients with XP, including close-ups (e.g. using a digital epiluminescence microscope). Pre-malignant lesions, such as actinic keratosis, should be treated in patients with XP according to the standard therapy guidelines (cryotherapy, curettage, or topical application of 5-fluorouracil). Interestingly, studies showed good therapy success of an imiquimod-containing cream in patients with XP (17). If invasive tumors are detected in the course of early skin cancer detection, the standard treatment is surgery. This should ideally be performed by an experienced surgeon, e.g. plastic surgeon, in order to keep the excision as small as possible. Another innovative approach to therapy is the use of skin grafts. In this case, skin is transplanted from UV-protected body sites of the patient (for example the thigh) being less likely to form tumors than the surrounding non-transplanted skin (18). In addition, therapeutic dermabrasion can be performed, in which the UV-damaged superficial skin layers are removed.

Summary of suggested clinical and molecular diagnostics in xeroderma pigmentosum involving several clinical specialities and state-of-the art molecular testing.
In some patients, high-dose oral therapies with isoretinoin can reduce the number of skin tumors occurring. However, since this therapy is accompanied by strong side-effects, such as hyperlipidemia, teratogenicity or calcification of ligaments and tendons, it should be used only in severely affected patients with a particularly high number of newly developed tumors (19, 20).
Furthermore, the topical application of xenogenic repair enzymes in cream form (photolyase or T4 endonuclease) has been shown to be quite effective as additional skin cancer prophylaxis in patients with XP (21, 22). Although daily use did not cause side-effects in patients, the treatment method is not yet approved for clinical use. Nonetheless, there are photolyase-containing sun screens, available from pharmacies, showing beneficial effects (23).
Recently, vismodegib, an oral inhibitor of the hedgehog signaling pathway, showed promising results in an 8-year-old patient with XP, leading to complete clearance of a nodular basal cell carcinoma on the tip of the nose (24). Additionally, anti-programmed cell death protein 1 therapy was very effective in inducing a regression of melanoma metastases and also non-melanoma skin cancer, especially in metastatic basal cell carcinoma with amplification of programmed cell death 1 ligand 1 (25, 26). This indicates that checkpoint inhibitor therapy is effective against all tumor entities (melanoma, basal and squamous cell carcinomas) with a high (sun-induced) mutational load.
There are innovative, but controversial attempts at gene therapy in patients with XP. In one approach, gene correction of primary keratinocytes was investigated to correct XP-C-deficient cells (27). Another attempt was to apply genome-editing tools, such as TALEN and meganucleases, to correct a mutation in an XP-C patient cell line (28). Although leading to significant improvement on the cellular level, these studies have not yet reached the clinical level. Gene therapy has several disadvantages that still have to be overcome, including the ex vivo treatment of patient cells after taking skin punch biopsies, outgrowing of the cells, modification and effective re-transplantation or delivery options (29). So far, there have been no gene therapy trials applying the CRISPR/Cas9 technique, but this innovative system could be engineered for efficient genetic modification in the future (30). Our group was able to generate a model cell line containing a complete knockout of the XPF gene, being a great tool to further clarify and investigate the different roles of XP proteins and give in-depth understanding of the disease and the complex genotype–phenotype correlations (29).
In many countries, patients and their families are organized in self-help groups, e.g. in the US, the Xeroderma Pigmentosum Society, which is dedicated to education and guidance (www.xps.org). In Europe, there are groups in Great Britain (joomla.xpsupportgroup.org.uk), France (http://www.enfantsdelalune.org/) and Germany (www.xerodermapigmentosum.de/). Various useful websites are available to assist physicians and scientists, as well as patients. Among others, there is a list of various disease-causing mutations (http://www.uniprot.org) as well as an international interest group, the DNA Repair Interest Group (http://sigs.nih.gov/DNA-repair/Pages/default.aspx).
Additional information and similar content on XP can also be found in further literature (4, 5, 29).
In conclusion, the consequences of defective NER are vividly demonstrated by the DNA repair-defective syndrome XP. Patients suffering from XP and reduced DNA repair in their skin develop melanoma, basal and squamous cell carcinoma. This demonstrates the role of UV-induced DNA damage as the tumor-driving cause. Both phenotypes and genotypes can be quite variable. This renders clinical, as well as molecular-genetic diagnostics challenging. To this end, the European Union has launched a program and established a European Reference Network Center for Rare Skin Diseases, including DNA-repair-defective disorders, in order to homogenize patient care, education, and research on new treatments (https://webgate.ec.europa.eu/ern/).
Acknowledgements
The Authors would like to thank German Cancer Aid (Deutsche Krebshilfe e.V.) [111377], the Claudia von Schilling Foundation for Breast Cancer Research, and the Heinz and Heide Duerr Foundation [2014-2.2.1/02] for funding of their research.
Footnotes
↵* These Authors contributed equally to this article.
- Received November 1, 2017.
- Revision received November 30, 2017.
- Accepted December 4, 2017.
- Copyright© 2018, International Institute of Anticancer Research (Dr. George J. Delinasios), All rights reserved





{kind=link}
{kind=link}