


Abstract
Background/Aim: Colorectal cancer (CRC) is the third most common cancer in the world. The high mortality of this tumor is mainly due to its invasive properties, as it forms metastases in multiple organs, preferentially in the liver. There has evidence showing that C-X-C chemokine receptor type 4 (CXCR-4) and its ligand, stromal cell-derived factor-1 (SDF-1), plays an important role in cancer progression and metastasis. However, the molecular mechanism underling the CRCR4-mediated CRC metastasis has not been well characterized. In this study, we aimed to investigate the roles of CXCR4 in colorectal cancer using the clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9 (Cas9)-based genomic editing technique. Materials and Methods: We knocked-down CXCR4 using specific guide-RNA linked CRISPR/Cas9 in HT115 and COLO201 colon cancer cell lines which exhibited high levels of endogenous CXCR4 gene expression. Stable HT115 cells with CXCR4 knock-down were established by CRISPR plasmid transfection and validation was confirmed using T7 endonuclease 1 (T7EN1), flow cytometry (FACS) and western blotting assays. Results: Knock-down of CXCR4 did not decrease proliferation of HT115 cells, but decreased the adhesion potential of cells to the human umbilical vein endothelial cells (HUVEC) and extracellular matrix. We further demonstrated that the AKT and type 1 insulin-like growth factor receptor (IGF1R) signalling pathways may be involved in the alteration of adhesion in CRC cells when CXCR4 is knocked down. Conclusion: Our data suggest that CXCR4 plays a key role in colorectal cancer progression via the mediation of tumor cell adhesion.
Colorectal cancer (CRC) is the third most common cancer in the world, with nearly 1.23 million new cases diagnosed every year (1). In Europe, CRC is the second leading cause of cancer-related death (2). The high mortality of this tumor is mainly due to its invasive properties, with distant metastasis in 75% of cases, especially to the liver (3). The survival-rate for patients without metastasis is 64% and it decreases to 12% when the malignancy spreads into other organs (4). Metastasis progression can be viewed as a stepwise sequence of events that include local invasion within the same organ, intravasation into blood or lymph vessels, survival of the malignant cells in the circulation, homing and extravasation and finally the colonisation of new tissues. The anatomical layout of the vasculature could partly explain liver metastasis of CRC, as the portal venous system drains most CRC cells into the liver (5). With similar vasculature, death from advanced gastric cancer is frequently caused by peritoneal carcinomatosis (6). The well-established “seed and soil” theory explains the formation of the metastasis in all tumors, including colorectal cancer, however the molecular mechanisms have not yet been elucidated (7).
C-X-C chemokine receptor type 4 (CXCR4) is one of the members of the chemokine, G-protein–coupled transmembrane receptors and is also named 7- transmembrane domain receptorss due to the 7-transmembrane helices adjacent to their N-terminal extracellular domain. Together with its ligand, the chemokine stromal cell-derived factor-1 (SDF-1), also named CXCL12, is released by the stromal cells, where it triggers various downstream signalling pathways that result in many responses, such as increase in intracellular calcium, gene transcription, chemotaxis, cell survival and proliferation. The signalling cascade induced by CXCR4 and its ligand is commonly defined as the “CXCR4’SDF-1 axis” and was first described in the trafficking and homing of lymphocytes in inflammation (8). However, the involvement of CXCR4 has also been demonstrated in many cancers. In particular, CXCR4 has been found to be up-regulated in more than 23 malignancies, including breast cancer (8, 9), colorectal cancer (9-12), lung cancer (9, 13) and prostate cancer (9, 14). Some studies have suggested that it has a role in the spread of tumors to distant sites (8).
In CRC, the higher expression of CXCR4 has been found to correlate with the stage of the disease and poor survival of patients (9), with higher levels in advanced TNM stages and when the tumor has spread into lymph nodes and liver (10-12).
The CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) and CRISPR-associated (Cas) genes have been found to be involved in the immune response of many bacteria and archaea and this system can now be used to engineer eukaryotic cells for the study of several diseases, including cancer. Cas9 is a nuclease that promotes the cleavage of a genomic locus by stimulating the DNA double-strand break repair (DBS), thus removing the target sequence from the genome. The CRISPR/Cas9 system has been demonstrated as a highly specific and efficient genetic editing tool compared to other gene silencing approaches such as siRNA or shRNA (13-15).
In the present study, we knocked down CXCR4 using a CRISPR/Cas9 system to investigate the role of CXCR4 in CRC cell lines. We also studied the effect of CXCR4 deletion on different pathways involved in colorectal cancer progression.
Materials and Methods
Human colorectal specimens. A total of 364 patient tissues (150 paired colorectal cancer tissues with adjacent normal tissues, 186 colorectal cancer tissues, 28 normal tissues) were collected immediately after surgery and snap-frozen in liquid nitrogen until further use. The size of tumor tissues and normal tissues was confirmed by a pathologist and the background tissues were free of tumor deposits. All protocols were approved by the Beijing Friendship Hospital Research Ethical Committee. Written consents were obtained from the patients. Full details of patient clinical data are shown in Table I.
Cell culture. The colorectal cancer cell lines including HT115, LS174T and COLO201 were obtained from the American Type Culture Collection (ATCC, Rockville, Maryland, USA). They were routinely cultured in DMEM/F12 HAM (Dulbecco's modified Eagle's medium; Sigma-Aldrich) supplemented with 10% foetal calf serum (FCS; PAA Laboratories, Somerset, UK) and 1x penicillin and streptomycin (Sigma-Aldrich, Poole, UK). All the cells were cultured in an incubator at 37.0°C with 95% humidity and 5% CO2. The recombinant human SDF-1 protein was purchased from PeproTech (London, UK) and used at the concentration of 50 ng/ml.
RNA isolation and RT-PCR. RNA was isolated from the colorectal cancer cells using total-RNA isolation reagent (Sigma-Aldrich, Dorset, UK). Following the manufacturer's protocol, cDNA was generated from the total RNA using GoScript™ Reverse Transcription System kit (Promega, Madison, WI, USA). PCR was conducted using a REDTaq™ ReadyMix PCR reaction mix. Real-time PCR gene expression analysis results were analysed using 2-(ΔΔCT) method of normalisation to the housekeeping gene GAPDH. All the primer sequences are shown in Table I.
CXCR4-gRNA-Cas9 vector construction. The CRISPR/Cas 9 vector was based on the pSpCas9 (BB)-2A-Puro (PX459) backbone plasmid (#48139, Addgene, Cambridge, MA, USA). Following the protocol described previously (16), five primers of the CAS9/guide RNA (gRNA) specific to CXCR4 were designed (Table I), annealed and inserted into the PX459 which was pre-digested with BbsI (R0539L; New England Biolabs, Ipswich, MA, USA). The constructed vectors were then transformed into a competent Escherichia coli strain for plasmid amplification in the presence of ampicillin. The vector DNA isolated from a single colony was then validated by sequencing using the LKO.1 5’ primer which is located upstream of the gRNA site in the vectors (Table I).
Electroporation. Cancer cells were electro-transfected with constructed CRISPR/Cas 9 plasmids or control plasmids. In brief, 1×106 HT115, COLO201 cells per sample were collected and washed twice in PBS. The cells were re-suspended in 800 μl serum-free medium with 2 μg of plasmids respectively. The cell/DNA mixture was transferred into the certified cuvette and electro-transfected with a Gene Pulser Xcell™ system (Bio-Rad, Hercules, CA, USA). After transfection, the cells were cultured in DMEM or RPMI 1640 medium supplemented with 10% FCS (without antibiotics) for 24 h, then changed to DMEM or RPMI 1640 medium with 10% FCS and antibiotics. After 48 h, cells were selected with 2 ng/ml puromycin for 7 days, which was continued with 0.2 ng/ml puromycin for maintenance.
T7 endonuclease I assay. The genomic DNA was extracted from cells using a Quick-DNA™ Universal Kit (Zymo Research Europe, Germany) according to the manufacturer's protocol. T7 endonuclease I assay (T7EN1) was performed following a protocol from fly CRISPR/Cas9 experiments, German Cancer Research Center (DKFZ). Briefly, the genomic region flanking the gRNA target site was PCR amplified using CXCR4 sequence primer (Table I). The PCR products were then purified, followed by a T7 endonuclease I digest. The mutation frequency was quantified by Image J software (National Institutes of Health, New York, USA) and calculated as described previously (17).
Western blot and flow cytometry (FACS). Equal amounts of proteins were separated using SDS–PAGE and blotted onto PVDF membrane. Proteins were then separately probed with a polyclonal rabbit anti-human CXCR4 antibody (Santa Cruz, Dallas, Texas, USA), monoclonal mouse anti-human GAPDH antibody (BD Biosciences, San Diego, CA, USA) and corresponding peroxidase-conjugated secondary antibody. Protein bands were visualised using a chemiluminescence detection kit (EZ-ECL, Biological Industries, Beit-Haemek, Israel) and photographed using a G:Box (Syngene, Cambridge, UK).
Primer sequence.
To determine CXCR4 disruption efficiency, the transfected cells were stained with monoclonal mouse anti-human CXCR4 antibody (Santa Cruz, Dallas, Texas, USA) and analysed or sorted by flow cytometry (FACS Canto II, BD biosciences, San Diego, CA, USA). The data were plotted in FCS Express 4 (De Novo Software, Thornhill, ON, Canada).
AlamarBlue® cell proliferation assay. Cell suspensions were added into a black 96-well plate (5,000 cells/100 μl/well). Cell growth was assessed at different time points (0, 24, 48, 72, 96 h) and in quadruplicate, 10 μl of AlamarBlue® Cell Viability Solution (ThermoFisher UK Ltd., Hemel Hempstead, UK) was added to each well. The plate was incubated at 37°C in the dark for four hours and then read on a GLOMAX® MULTI Detection System (Promega, Southampton, UK) at 570 nm in relative fluorescence units.
Tumor-endothelium adhesion assay. HUVEC cells were obtained from LONZA ((Lonza, Slough, UK) and maintained in EGM2 medium (Lonza, Slough, UK). HUVEC cells were seeded into a 48-well plate at a density of 3×105 cells/well and allowed to form a monolayer. Cancer cells were diluted to 1×106 cells/ml and 5 μl/ml of Vybrant DiO cell-labelling solution (ThermoFisher UK Ltd., Hemel Hempstead, UK) were added. Cells were incubated for 20 min and then washed three times using PBS. Cells were resuspended at 2×105 cells/ml and 200 μl added to the 48-well plate containing HMVEC monolayer. Cells were allowed to attach for 30 min, and then washed twice with PBS following which, 350 μl of cell dissociation solution was added to each well and the plates further incubated for 1 h. The cell suspension was aliquoted into a black 96-well plate and read on a GLOMAX® MULTI Detection System, at 495 nm excitation and 519 nm emission.
Matrigel adhesion assay. A total of 20,000 cells were added to each well of a 96-well plate which was pre-coated with Matrigel (5 μg/100 μl/well) (Corning, BD, UK). After 40 min of incubation, non-adherent cells were washed off using BSS buffer. The remaining cells were fixed with formalin and stained with crystal violet. The number of adherent cells was then counted under a microscope.
Transwell migration assay. Cells were harvested and resuspended in serum-free medium, at a density of 1×105 cells/ml. DMEM (1ml) containing 10% FCS (chemoattractant) was added to the receiver wells in triplicate, and 1ml of serum-free DMEM (no chemoattractant) was added to the receiver well of the control transwell. An 8-μm-pore ThinCert™ 24-well plate insert (Greiner Bio One, Frickenhausen, Germany) was placed in each of the receiver wells and 500 μl of cell suspension was added to each transwell insert. Following four hours of incubation, the transwell inserts were washed gently with PBS and then incubated for a further hour in 350 μl cell dissociation solution (CDS; Sigma–Aldrich, Irvine, UK) containing Calcein AM (eBioscience, Hatfield, UK) at a ratio of 1.2 μl Calcein AM (1.67 μg/μl) in 1 ml 1X CDS. Following this, the cell suspension was aliquoted into a black 96-well plate and read on a GLOMAX® MULTI Detection System, at 495 nm excitation and 519 nm emission. To analyse the total directed cell movement, the fluorescence of the well containing no chemoattractant was subtracted from that of the test wells.
Transwell invasion assay. Matrigel was added to serum-free medium to gain a concentration of 500 μg/ml, then 100 μl of this solution was added to 8-μm-pore ThinCert™ 24-well plate inserts and allowed to incubate for 1 h at 37°C. The rest of the invasion assay was undertaken in the same way as the migration assay, except the running time of the experiment was 24 h.
Electric cell-substrate impedance sensing (ECIS). An ECIS system utilising 96-well array plates (Applied Biophysics, Inc., Troy, NY, USA) was used to quantify cell behaviour. Briefly, cells were prepared with six repeats per group and seeded at 30,000 cells/well in 200 μl of complete culture medium. The corresponding values of electrical impedance between the electrodes due to the alteration of the cellular adhesion and motility properties in an ECIS array were recorded and analysed in this system.
Statistical analysis. A Shapiro-Wilk test was used to verify if the data were normally distributed. T-tests were used for parametric distributions. Mann-Whitney and Wilcoxon Signed-Rank Tests were used for non-parametric distributions. Statistical analysis was performed using GraphPad Prism 6.0 software (GraphPad Software, San Diego, CA, USA. Statistical significance was indicated with the following nomenclature: *p<0.05, **p<0.01, ***p<0.001.
Gene expression of CXCR4 in the CRC cohort.
Results
CXCR4 gene expression in colorectal cancer tissues and cell lines. As shown in Table II, in the tissues of colorectal cancer patients the expression of CXCR4 varied according to the grade of the tumor, with a lower expression in the cancer tissues compared to their normal counterparts (p=0.0234) and the lowest levels in the metastatic group (p=0.0342). The expression of CXCR4 was also investigated in different cell lines and its expression was found to be high in HT115 and COLO201 cells (Figure 1A and B). The HT115 cell line was chosen as the model in this study of CXCR4 as its expression was shown to be very high in these cells.
CRISPR/Cas9-knock-down CXCR4 model in colorectal cancer cell lines. In order to knock-down CXCR4 efficiently by using the CRISP/Cas9 technique, five gRNAs were designed to target CXCR4 (G1-G5) and the control plasmid PX459 (backbone plasmid only). Two were finally chosen (G2 and G3), as they were the only ones to show the correct sequence. After selection of the successfully transfected HT115 cells, we verified that G3 was the only construct able to induce a small insertion and deletion (INDEL) mutation in the CXCR4 gene (Figure 2A).
In order to construct the CXCR4-gRNA-Cas9 vector, 5 gRNAs were designed to target Cas9. After Sanger sequencing, it was observed that G2 and G3 retained the correct sequence. We then electro-transfected the HT115 cell line with constructed CRISPR/Cas9 plasmids (G2, G3) and control plasmids (PX459). Approximately 14 days after the transduction and selection, we performed T7EN1 assays to measure CXCR4 DNA levels (Figure 2). After the selection of the HT115 cells that were successfully transfected, we verified that G3 was the only construct able to induce small insertions and deletions (INDELs) in the CXCR4 gene (Figure 2A). To further evaluate the efficiency of the CXCR4 disruption in HT115 cells mediated by the G2 and G3 plasmids, the protein level of CXCR4 was investigated by FACS and western blot (Figure 2). FACS data showed that the CXCR4 protein significantly decreased in the cells containing the G3 vector, whereas no significant differences were observed in the cells transfected with the control and G2 vectors (Figure 2C). The western blot results confirmed the FACS analysis (Figure 2B).

Expression of CXCR4 in colon cancer cells. (A) The mRNA levels of CXCR4 in colon cancer cell lines in HT115, LS174T, COLO201 cell lines. (B) CXCR4 mRNA levels were highly expressed in HT115 and COLO201 cell lines.
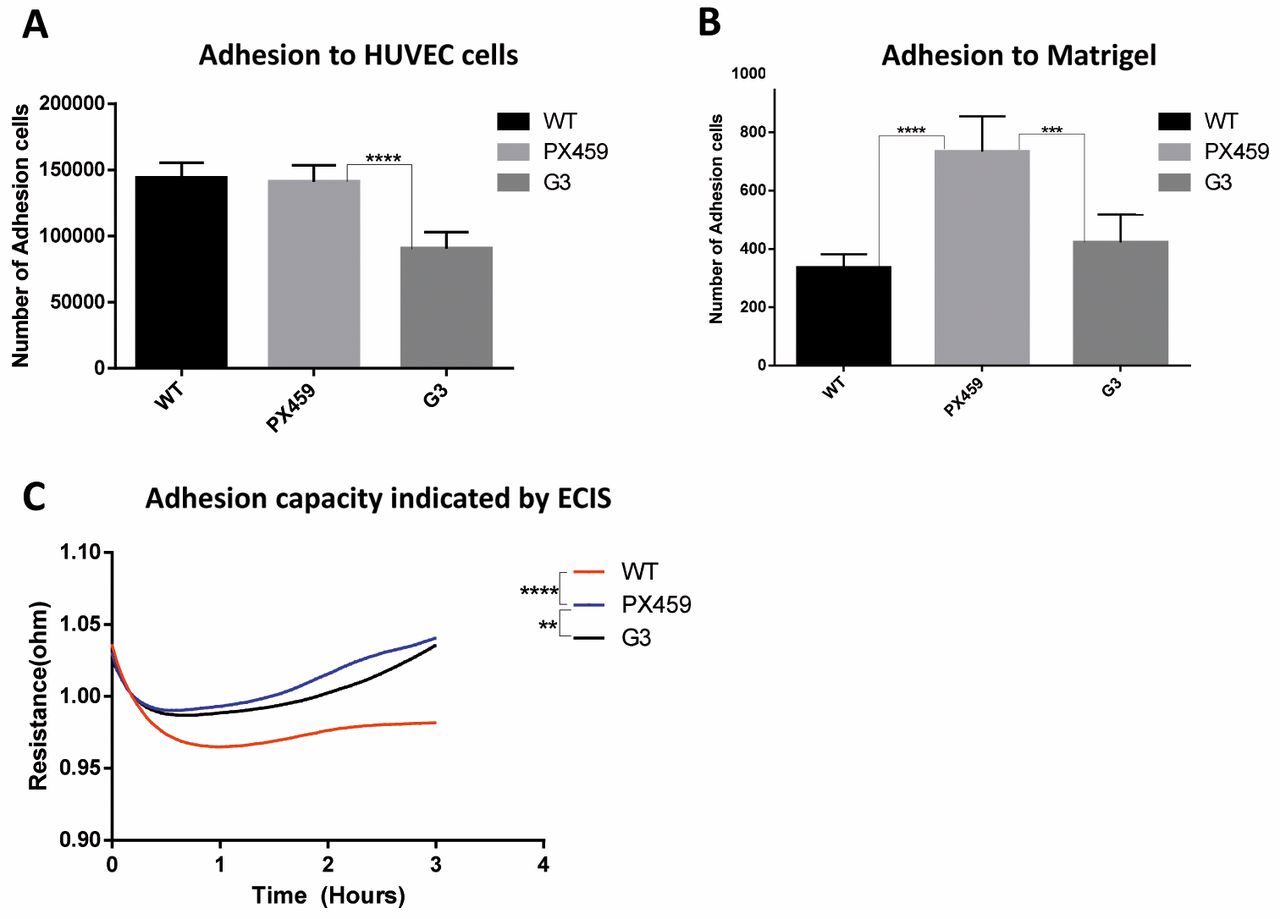
Effect on colorectal cancer cell functions by knock-down of CXCR4. The knock-down of CXCR4 mediated by the G3 construct significantly increased the proliferation of the HT115 cells after 4 days (p<0.001), whereas no differences were observed in the wild-type cells and cells transfected with the backbone plasmid only, at the same time point (Figure 3). We also investigated the effect of CXCR4 knock-down on the adhesive ability of the HT115 cells in vitro. The adhesion of the colon cancer cells to the extracellular matrix, reproduced with the use of the Matrigel, decreased significantly in the CXCR4-knock-down cells (containing G3 vector) compared to the cells with the backbone vector (p<0.001). However, we also observed the increase of adhesion to Matrigel from the backbone compared to the wild-type (Figure 4B). A similar effect was also seen when the HT115 cells were co-cultured with the primary endothelial cells HUVECs. In this latter case, the adhesive properties of colon cancer cells did not change when transfected with the backbone plasmid, whereas this decreased when CXCR4 was knocked-down (p<0.001; Figure 4A). The reduction of the adhesive capacity by the knock-down of CXCR4 in HT115 cells was confirmed by the ECIS system (p<0.01 vs. PX459; Figure 4C).

Knock-down of CXCR4 in the HT115 cell line. (A) T7EN1 assay of each gRNA CRISPR/Cas9 targeting CXCR4 (G1, G2 and G3) in HT115 cells. (B) FACS analysis of CXCR4 expression in HT115 cells. (C) Western blot analysis of CXCR4 expression in HT115 cells transduced with CXCR4-gRNA CRISPR/Cas9 vectors. The HT115-WT, -PX459 and -G3 cell lines were collected and analyzed by western blotting with an anti-CXCR4 or anti-GAPDH antibody.
We also determined the influence on cell migration and invasion. Knock down of CXCR4 showed no significant difference in migration and invasion of HT115 cells (Data not shown).
CXCR4 signalling and treatment with SDF-1. Recent studies have suggested that the CXCR4/SDF-1 axis triggers the activation of other pathways and molecules, including MEK1/2, ERK1/2 and PI3K-AKT. For this reason, we investigated the effect of knock down of CXCR4 on ERK1/2 and its upstream molecule AKT. Western blot results showed a slightly lower expression of ERK1/2 in the cells transfected with the G3 plasmid, compared to the wild type cells and cells containing the backbone plasmid only (Figure 5A). On the other hand, both the inactive and active forms of AKT (AKT1 and pAKTSer473) were significantly down-regulated in CXCR4 knock down HT115 cells (Figure 5B and C).

The effects of CXCR4 knock-down on proliferation of HT115 cells. Knock-down of CXCR4 increased the in vitro growth of HT115 cells on Day 2 and Day 3. *p<0.05.

Effect of CXCR4 knock-down on the adhesion of the HT115 cells. (A) Knock-down of CXCR4 decreased the in vitro tumor cell adhesion to the HUVEC cells (vs. backbone control). (B) Knock-down of CXCR4 decreased the in vitro tumor cell adhesion to Matrigel (vs. backbone control). (C) Knock-down of CXCR4 decreased the in vitro adhesion property of the HT115 cells as indicated by the Electric Cell-substrate Impedance Sensing (ECIS) system (vs. backbone control). **p<0.01; ***p<0.001; ****p<0.0001.
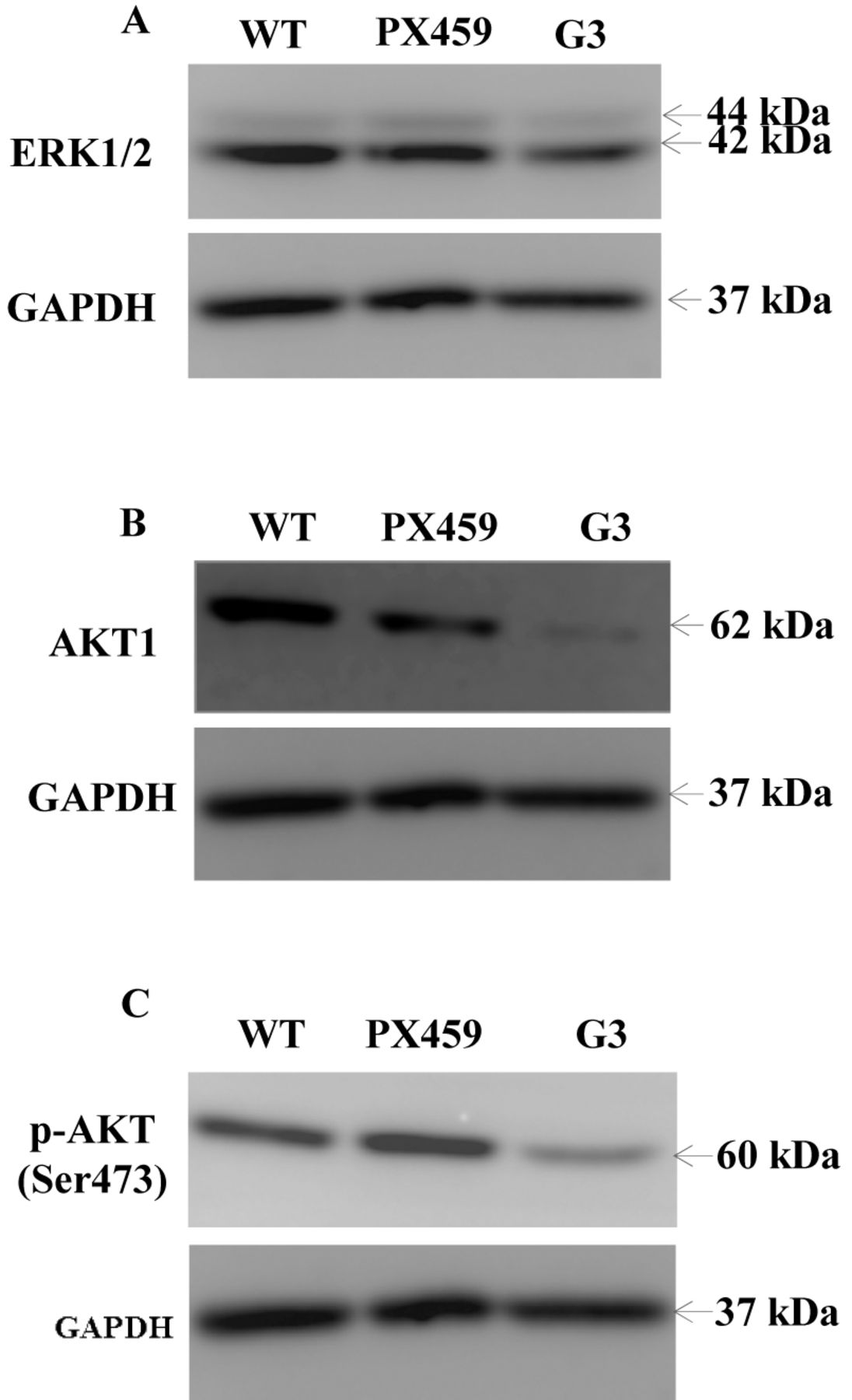
The effects of the CXCR4 knock-down on ERK1/2 and AKT signalling pathways in the HT115 cells. (A) Knock-down of CXCR4 cells slightly decreased ERK1/2 expression. (B) Knock-down of CXCR4 cells significantly decreased AKT1 expression. (C) p-AKT expression were significantly decreased in the CXCR4-knock-down HT115 cell line.
We also analysed the gene expression of proteins upstream of ERK1/2 in CXCR4 knock down HT115 cells treated with SDF-1. As shown in Figure 6, in the absence of SDF-1 treatment, CXCR4 knock-down led to the up-regulation of ERK, MEK, mTOR and IRF1R genes. However, after post-treatment with SDF-1 for 2 h, it appeared that the mRNA levels of ERK and MEK were both up-regulated in all cell groups, thus showing no significant difference. Interestingly, although post-treatment of the cells with SDF-1 also increased the level of mTOR gene expression, there was significant increase of mTOR gene expression in the CXCR4 knock-down cells in the presence of SDF-1 compared to the same cell line without SDF-1 (p<0.01). Although SDF-1 treatment increased the expression of IGF1R in the WT and PX459 cell groups, post-treatment of the CXCR4 knock down cells with SDF-1 reduced the IGF1R gene expression significantly (p<0.01).
Discussion
CXCR4 has been found to be up-regulated in colorectal cancer and its expression levels correlate with the stage of the tumor, lymph node and liver metastasis and with a higher expression in the most advanced stages of the disease. In our study, we verified that this is the case in tissues, but not in all the colorectal cancer cell lines, thus confirming there is a difference in the expression of CXCR4 in vitro and in vivo as shown in a previous study (18).
In our study, we successfully reduced the CXCR4 expression in the colorectal cancer cell line HT115 in a very specific manner. When CXCR4 was knocked down, there was an increase in the proliferation of the cancer cells, as well a decrease of the adhesion of the same cells on the primary endothelial cells (HUVECs) and at the same time, an increase of the adhesion ability of the HT115 to the extracellular matrix. Taken together, our data suggest that knock down of the SDF-1 receptor decreases the progression of the malignancy.
In some previous studies, CXCR4 was knocked down in colorectal cancer using siRNA silencing. One study showed a reduction of the proliferation, migration and invasion properties of HT-29 cells when CXCR4 was silenced (19). On the other hand, using the same system, another study did not find any difference in the proliferation of Caco-2, HT-29 and SW480 cell lines (20). The discrepancies between these studies may be due to the cellular difference or silencing efficiency. In our study, we used CRISPR/Cas9 knock-down CXCR4 in HT115 cells, revealing that knock down of CXCR4 cells increased cell proliferation but decreased cell adhesion. In vitro assay results provided evidence that CXCR4 decreased cell proliferation, which may be associated with lower expression of CXCR4 in the CRC tissue than normal tissue. In vitro adhesion assay showed CXCR4 involved in cell adhesion, suggested that the metastatic potential of CRC cells may be associated with expression of CXCR4.
Considering the recent findings on the pathways influenced by CXCR4-SDF-1 activation, we investigated the effect of the CXCR4 knock-down in colorectal cancer cells on PI3K/AKT/mTOR and RAF/MEK/ERK at the gene and protein level. At the gene level, after treatment with SDF-1 for 2 h, CXCR4 knock-down led to an increase of the ERK and MEK mRNA in the wild-type cells. On the other hand, knock-down of CXCR4 also resulted in up-regulation of these genes. However, there was no synergetic effect between CXCR4 knock-down and SDF-1 post-treatment. This finding is in line with previous reports indicating that the SDF1-/CXCR4 axis is involved in invasion and proliferation of certain tumor cells through ERK and MEK signalling pathways (21-24). In contrast, CXCR knock down and SDF-1 treatment showed a synergetic effect on the up-regulation of mTOR gene. Whereas SDF-1 post-treatment reduces the increased IGF1R gene expression by CXCR4 knock down. We also verified the protein levels of AKT, that is upstream of mTOR and regulated by ERK/MET, in CXCR4 knock-down cells without any treatment (25). We found that both the inactive and active forms of AKT are down-regulated when CXCR4 is knocked-down. This may suggest that the reduction of AKT leads to an increase to the mTOR levels in the cells when the CXCR4-SDF1 axis is suppressed and this involve both the RAF/ERK/MEK and PI3K/AKT/mTOR pathways.
As the disruption of the CXCR4/SDF-1 axis decreased the proliferative properties of colorectal cancer cells, we further explored the molecular mechanisms involved. In particular, we studied the expression of IGF1R, as some recent studies show an interaction between CXCR4 and IGF1R by binding in a complex (26). We found that the expression of IGF1R mRNA can be up-regulated by CXCR4 knock-down, which can be subsequently reduced by the post-treatment of SDF-1. This finding supports our hypothesis that IGF1R has a functional RTK/GPCR hybrid (27), and is therefore involved in CRC cell proliferation (28). This also indicates that the influence of the CXCR4/SDF-1 axis in the proliferation of colorectal cancer cells may be mediated by IGF1R. In conclusion therefore, with regard to potential clinical impact, knock-down of CXCR4 may suppress tumor-associated angiogenesis promoted by IGF1R in the presence of SDF-1 in the tumor microenvironment.
Footnotes
Conflicts of Interest
The Authors declare no conflicts of interest.
- Received May 19, 2017.
- Revision received June 6, 2017.
- Accepted June 7, 2017.
- Copyright© 2017, International Institute of Anticancer Research (Dr. George J. Delinasios), All rights reserved





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}