


Abstract
Aim: To investigate the molecular mechanisms by which long-term exposure to cigarette smoke extract (CSE) contributes to ovarian cancer metastasis. Materials and Methods: Western blot analysis for diverse p110 isoforms of phosphoinositide 3-kinase (PI3K)-related signaling pathway and epithelial-mesenchymal transition (EMT) markers was performed to analyze the underlying mechanisms. Migratory activity of CSE-exposed ovarian cancer cells was determined by transendothelial migration and invasion assay. Results: After exposure to CSE for four weeks, CaOV3 (primary) and SKOV3 (metastatic) ovarian cancer cells showed enhanced mesenchymal characteristics and produced EMT-related cytokines [intwerleukin-8 (IL-8), vascular endothelial growth factor (VEGF) and transforming growth factor-beta 1 (TGF-β1)]. CSE exposure activated the Src-p110δ-p21 protein-activated kinase 1 (PAK1) in CaOV3 cells and the Lyn-p110β-Rho-associated kinases 1/2 (ROCK1/2) in SKOV3 cells, which led to the stimulation of LIM kinase 1 (LIMK1) phosphorylation and TGF-β1 release. LIMK1 knockdown efficiently blocked the migratory activity and suppressed the mesenchymal phenotypes of CSE-treated ovarian cancer cells. Reactive oxygen species (ROS) initiated the CSE-mediated EMT processes in ovarian cancer cells. Conclusion: Characterization of the p110 isotypes of PI3K is critical for regulating cancer metastasis; LIMK1 could be a common therapeutic target of ovarian cancer metastasis.
- Phosphoinositide 3-kinase
- LIM kinase 1
- ovarian cancer
- p21 protein-activated kinase 1
- rho-associated kinases 1/2
- epithelial-mesenchymal transition
- cigarette smoke extract
Several studies have shown a strong correlation between cigarette smoke (CS) and cancer metastasis through the induction of numerous factors involved in migration activity (1-3). The exposure to CS induces the epithelial-mesenchymal transition (EMT) process and up-regulates the expression of EMT markers, including N-cadherin and vimentin (4, 5). Cigarette smoke extract (CSE) treatment significantly induces interleukin-8 (IL-8) and transforming growth factor-beta 1 (TGF-β1) production and profoundly suppresses the proliferation and growth of erythroid and granulocyte-macrophage progenitors (6). Stimulation with CSE in human lung fibroblast cells induces the expression of phosphorylated Smad3, a main downstream target of the TGF-β1 receptor, which results in the secretion of vascular endothelial growth factor (VEGF) (7). CSE increases the migratory and invasive activity of ovarian cancer cells and induces the expression of the EMT-related transcriptional factors Snail and Slug (8). Nonetheless, the mechanisms underlying the induction of EMT by CSEs in ovarian cancer remain unclear.
TGF-β, a well-established EMT mediator, plays a critical role in tumor progression (9, 10). TGF-β1 activates the phosphorylation of Akt, which, in turn, induces the delocalization of E-cadherin and the acquisition of a spindle cell morphology (11). The activation of the phosphoinositide 3-kinase (PI3K)/Akt pathway increases Twist1 expression and sequentially up-regulates TGF-β2 secretion and TGF-β-mediated signaling. In addition, enhanced TGF-β receptor signaling maintains hyperactive PI3K/Akt signaling (12). The precise mechanism by which the PI3K/Akt signaling pathway reinforces TGF-β1 production remains controversial.
PI3K and Akt have a wide influence on several cellular processes, such as cell growth, survival, migration and metabolic control (13). Class I PI3Ks are the best-characterized enzymes, including class IA (p110α, p110β and p110δ) and class IB (p110γ), and the aberrant activation of this pathway has been widely reported in many human cancers, including ovarian cancer (14). Overexpression and mutations in the PIK3CA gene encoding the p110α catalytic subunit of PI3K, which is frequently activated, have been frequently detected in various cancers, including breast, ovarian, colorectal and liver (15-17). In addition, the expression of PI3K p110β isoforms is related to chemoresistance in ovarian cancer cell lines (18). However, the role of the specific isoforms of PI3K is currently unknown in CSE-mediated EMT processes and TGF-β1 production in ovarian cancer cells. Based on these results, targeting the particular PI3K isoforms in ovarian cancer may provide experimental information for effective therapeutic treatments.
The aberrant expression of Rho and downstream targets, Rho-associated protein kinase 1 (ROCK1) and ROCK2, is commonly observed in human cancers and often associated with more invasive and metastatic phenotypes (19). Rho is one of the downstream targets of PI3K and treatment with a PI3K inhibitor or ROCK inhibitor inhibits the transendothelial migration of mesenchymal stem cells (20, 21). Treatment with inhibitors of RhoA, ROCK1/2 and PI3K attenuates TGF-β1 expression (22). LIM kinase 1 (LIMK1), which is activated by p21-activated kinase (PAK) and ROCK, also contributes to cancer cell migration and invasion by regulating actin dynamics (23-25). TGF-β1 activates cytoskeletal actin rearrangement through Rho/ROCK-mediated LIMK1 activation causing a fibrotic response in retinal pigment epithelial cells (26). Meanwhile, the regulatory effect of LIMK1 on TGF-β1 production in ovarian cancer cells remains unclear.
In this study, we chose primary (CaOV3) and metastatic (SKOV3) epithelial ovarian cancer cell lines to characterize the altered expression of the p110 isoforms and define the target signaling molecules. We investigated the controlling mechanism of PI3K-mediated LIMK1 activation to enhance TGF-β1 production and EMT processes in CSE-exposed ovarian cancer cells.
Materials and Methods
Cell lines and chemicals. The human ovarian cancer cell lines CaOV3 and SKOV3 were purchased from the ATCC (Manassas, VA, USA). CaOV3 cells were maintained in DMEM medium (Corning Incorporated, Corning, NY, USA) supplemented with 10% FBS (RMBIO, Missoula, MT, USA), penicillin, streptomycin and glutamine at 37°C in 5% CO2. SKOV3 cells were maintained in RPMI-1640 medium (Corning Incorporated) supplemented with 10% FBS (RMBIO), penicillin, streptomycin and glutamine at 37°C in 5% CO2. TGX-221 (p110β inhibitor), CAL-101 (p110δ inhibitor), PP1 (Src/Lyn inhibitor), LY2109761 (TGF-β receptor type I/II inhibitor), RKI-1447 (ROCK1/2 inhibitor) and IPA-3 (PAK1 inhibitor) were obtained from Selleckchem (Houston, TX, USA). NAC (reactive oxygen species (ROS) scavenger) was purchased from Sigma-Aldrich (St. Louis, MO, USA).
Preparation of cigarette smoke extract (CSE). We obtained the research-grade cigarettes (3R4F) from the Kentucky Tobacco Research Council (University of Kentucky, Lexington, KY, USA). Each cigarette contained 0.726 mg of nicotine, 9.4 mg of tar and 10.9 mg of total particulate matter. CSE was an extract of mainstream cigarette smoke. Briefly, the smoke was bubbled through 25 ml of DMEM and RPMI-1640 medium without FBS for 2 min and this solution was used as the stock (100%) for further dilutions. After adjusting the pH to 7.2, the obtained CSE was filtered through a 0.22 μm pore filter (Millipore, Bedford, MA, USA) for sterilization. CSE was standardized by measuring the absorbance at a wavelength of 320 nm and was used within 30 min after the preparation (27).
Western blotting. The cells were washed in PBS and lysed in NP-40 buffer (Elpis Biotech, Daejeon, Korea) supplemented with a protease inhibitor cocktail (Sigma-Aldrich). Protein phosphorylation states were preserved through the addition of phosphatase inhibitors (Cocktail II, Sigma-Aldrich) to the NP-40 buffer. The protein concentrations were determined using a bicinchoninic acid (BCA) assay kit (Pierce, Rockford, IL, USA). The proteins (10 μg/sample) were resolved through sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and then transferred to a nitrocellulose membrane (Millipore Corp., Billerica, MA, USA). The membranes were blocked with 5% skim milk prior to Western blot analysis. Chemiluminescence was detected using an enhanced chemiluminescence (ECL) kit (Advansta Corp., Menlo Park, CA, USA) and the multiple gel documentation (Gel DOC) system (Fujifilm). The following primary antibodies were used: E-cadherin, N-cadherin, vimentin, ZO-1, snail, β-actin, p110α, p110β, p110γ, p110δ, phospho-PAK1 (Ser144), PAK1, phospho-PAK4 (Ser474), PAK4, RhoA, Fyn, phospho-Lyn (Tyr507), Lyn, phospho-Src (Tyr416) and Src (Cell Signaling Technology, Beverly, MA, USA); alpha(α)-smooth muscle actin (α-SMA) (Bioss, Woburn, MA, USA); ROCK1 and ROCK2 (Abcam, Cambridge, UK); and phospho-LIMK1 (Thr505), LIMK1 and phospho-Fyn (Thr12) (Santa Cruz Biotechnology, Santa Cruz, CA, USA).
Small interfering RNA (siRNA) transfection. An experimentally verified human LIMK1 small interfering RNA (siRNA) duplex and a negative control siRNA were obtained from Bioneer Corporation (Daejeon, Korea). The cells were seeded at a concentration of 1×105 per well in a 6-well plate and grown overnight. The cells were then transfected with 200 nM siRNA using LipofectAMINE RNAiMAX Reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions. The cells were used 48 h after transfection for further experiments.
Quantification of human cytokines by ELISA. The conditioned media were collected and active TGF-β1, VEGF and IL-8 were quantified using a single cytokine ELISA assay kit (R&D Systems, Minneapolis, MN, USA) according to the manufacturer's instructions. The data are expressed as the average of the number of biological replicates±standard deviation (SD).
Transendothelial migration and invasion assay. The transendothelial migration assay kit (Cell Biolabs, Inc., San Diego, CA, USA) was used according to the manufacturer's instructions 4 weeks after infection. The relative fluorescence units (RFU) of the migrated cells were measured using a microplate reader. Invasion was determined using the CultreCoat 96-well Medium BME Cell Invasion Assay Kit (R&D Systems) according to the manufacturer's protocol. The cells (2.5×104) in serum-free RPMI-1640 or DMEM containing 0.1% FBS were seeded into the upper chamber and the lower compartment was filled with RPMI-1640 or DMEM containing 10% FBS, which served as a chemoattractant. After incubation for 24 h, non-invading cells on the upper membrane surface were removed by wiping with a cotton swab. Invaded cells were stained with calcein-AM (Sigma-Aldrich) and quantified using a microplate reader.
Detection of intracellular ROS generation. The intracellular accumulation of ROS was examined by flow cytometry after being stained with the fluorescent probe, DCFH-DA (10 μM, 2’7’-dichlorodihydro-fluorescein diacetate; Molecular probes, Eugene, OR, USA). DCFH-DA was deacetylated in the cells by esterase to a non-fluorescent compound, DCFH, which remains trapped within the cell and is cleaved and oxidized by ROS in the presence of endogenous peroxidase to a highly fluorescent compound, DCF (2’7’-dichlorofluorescein). Briefly, the cells were seeded in 6-well plates (1×105 cells/ml), treated with or without CSE and incubated with 10 μM DCFH-DA for 30 min at 37°C. Then, the cells were washed and re-suspended in PBS and ROS levels were determined using an Accuri C6 flow cytometer (BD Biosciences, San Jose, CA, USA).
Statistical analysis. The data are expressed as the mean±standard deviation (SD). Statistical analysis was conducted using one-way analysis of variance. A p-value <0.05 was considered statistically significant.
Results
CSE induces the mesenchymal phenotype and invasive activity of ovarian cancer cells. We first investigated whether CSE stimulates the mesenchymal phenotype and morphology change in CaOV3 and SKOV3 cells. CSE supplementation for 4 weeks had no effect on cell growth and survival compared to the levels of the non-treated control group (data not shown). After culturing in the presence of CSE for 4 weeks, both ovarian cancer cells changed into mesenchymal spindle-type cells (Figure 1A). In addition, epithelial markers (E-cadherin and ZO-1) disappeared in a time-dependent manner, whereas the molecular markers (N-cadherin, vimentin, α-SMA, and snail) for mesenchymal cells increased during the first week after treatment with CSE (Figure 1B). The migratory activity and invasion ability were enhanced in CSE-treated ovarian cancer cells (Figure 1C) and the production of EMT-related cytokines, including IL-8, VEGF and TGF-β1, were significantly elevated after stimulation with CSE (Figure 1D). These results suggest that long-term exposure to CSE modifies the cancer cell into an aggressive phenotype that is capable of promoting metastasis.
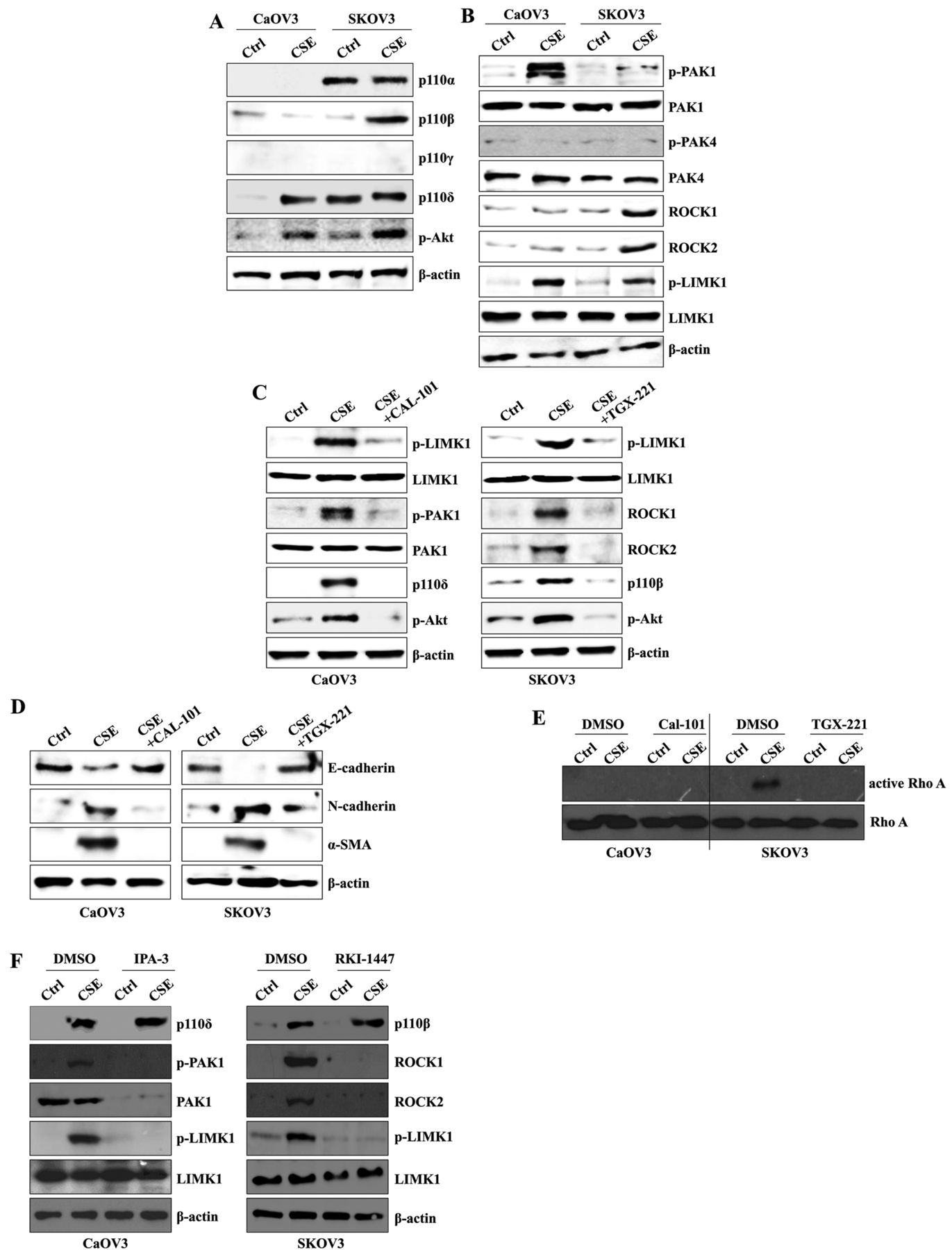
CSE exposure activates the p110δ-PAK1 pathway in CaOV3 cells and the p110β-ROCK1/2 pathway in SKOV3 cells. CSE induces TGF-β1 production through the activation of the PI3K/Akt pathway, which results in activation of the EMT process (28). PI3K/Akt signaling plays a critical role in Rho family signal transduction to contribute to cell migration (29). Based on these reports, we next examined whether long-term exposure to CSE involves the alteration or selective activation of a specific catalytic unit of class I PI3K to enhance the EMT process. Although CaOV3 cells displayed very low levels of PI3K before CSE stimulation, the expression of the p110δ isoform was significantly up-regulated in CSE-exposed CaOV3 cells (Figure 2A). Specifically, the p110α and p110γ isoform of PI3K were both identified in SKOV3 cells. The expressional level of p110β was profoundly elevated after long-term culture in the presence of CSE (Figure 2A). The expression of p110γ was not detected in CSE-treated and non-treated ovarian cancer cells (Figure 2A). Next, we analyzed the downstream target of PI3K to define the CSE-mediated EMT mechanism. The expression of phosphorylated PAK1 and ROCK1/2 was up-regulated in CSE-exposed CaOV3 and SKOV3 cells, respectively. Furthermore, the phosphorylation of LIMK1 was induced in both cell types treated with CSE for four weeks (Figure 2B). To clarify the role of PI3K in the stimulation of the downstream target molecules, CSE-exposed ovarian cancer cells were treated with a p110δ-specific inhibitor (CAL-101) or a p110β-specific inhibitor (TGX-221). The inhibition of p110δ blocked the expression of phosphorylated PAK1 and LIMK1 and the down-regulation of p110β reduced the activation ROCK1/2 and LIMK1 (Figure 2C). Treatment with CAL-101 and TGX-221 in CSE-stimulated ovarian cancer cells also reduced the expression of mesenchymal markers (Figure 2D). The pharmacological inhibition of p110β efficiently suppressed the expression of active Rho, while targeted inhibition of ROCK1/2 using RKI-1447 prevented the phosphorylation of LIMK1 in CSE-stimulated SKOV3 cells (Figure 2E and 2F). In addition, treatment with IPA-3 (PAK1 inhibitor) in CSE-treated CaOV3 cells prevented the activation of LIMK1 (Figure 2F). These results suggest that activation of the specific p110 catalytic isoform initiates the CSE-mediated EMT processes of ovarian cancer in a cell-type dependent manner.
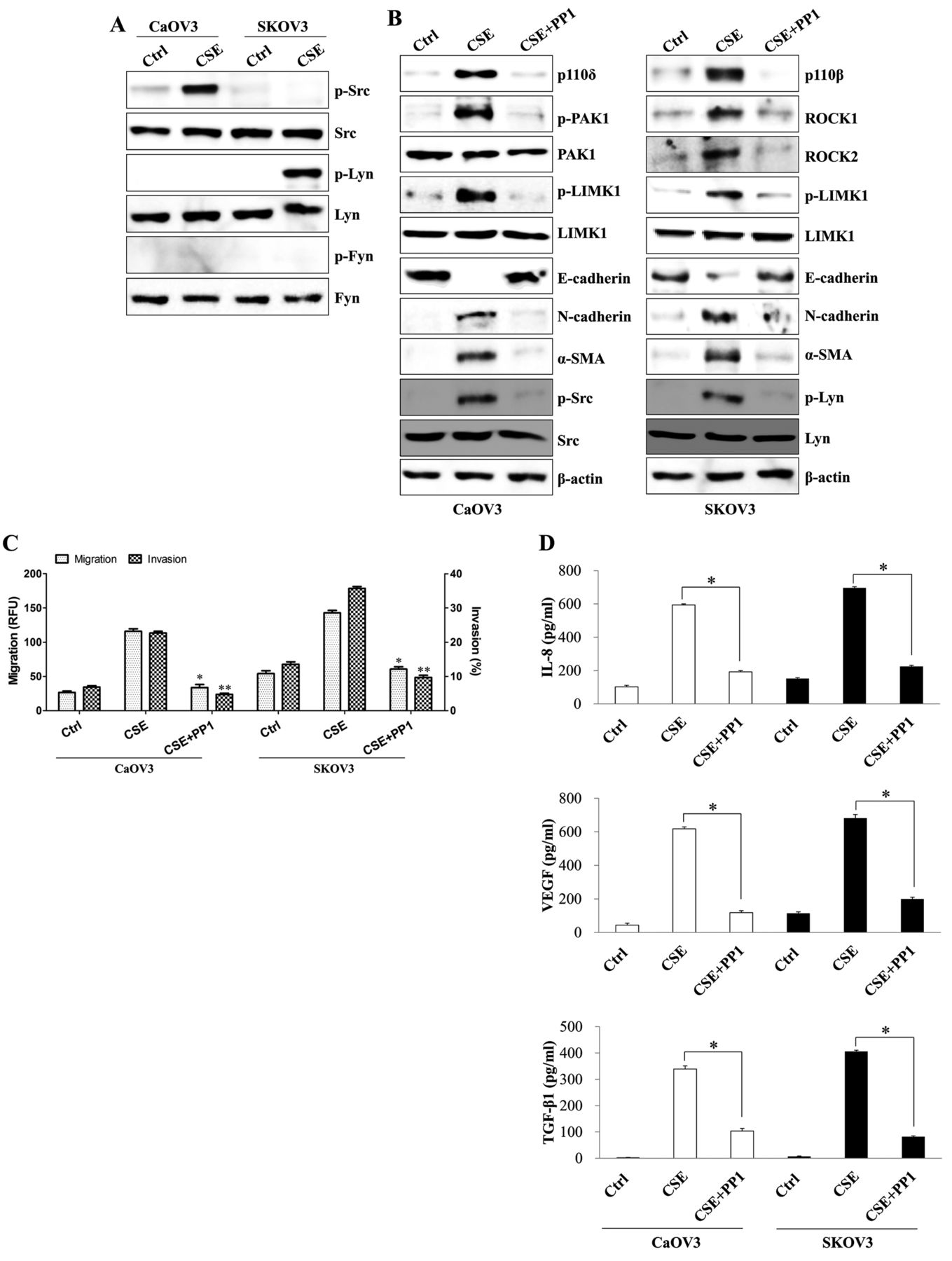
The Src family of kinases trigger PI3K-mediated EMT processes in CSE-exposed ovarian cancer cells. The Src tyrosine kinase family (Src, Lyn, and Fyn) modulates the PI3K signaling pathway (30, 31). Activation of Src kinase by CSE is critical for EMT induction (4). By contrast, Src kinase negatively regulates TGF-β1-dependent tumor invasion and metastasis of ovarian cancer cells, despite the activation of PI3K by that kinase (32). We next examined whether CSE-mediated EMT processes and TGF-β1 production are connected to Src-mediated PI3K activation. Long-term exposure to CSE resulted in activation of Src kinase in CaOV3 cells and Lyn kinase in SKOV3 cells (Figure 3A). Pharmacological inhibition of Src tyrosine kinase family using PP1 not only attenuated the expression of phosphorylated PAK1, phosphorylated LIMK1 and mesenchymal markers but also recovered the expression of E-cadherin in CSE-treated CaOV3 cells (Figure 3B). In addition, treatment with PP1 in CSE-exposed SKOV3 cells blocked the phosphorylation of ROCK1/2 and LIMK1, as well as the expression of N-cadherin and alpha-smooth muscle actin (α-SMA) (Figure 3B). PP1 efficiently suppressed the migration and invasion activity of CSE-stimulated ovarian cancer cells and the production of EMT-related cytokines (Figure 3C and D). These results suggest that Src-induced PI3K activation controls PAK1- or ROCK1/2-related EMT processes in CSE-exposed ovarian cancer cells.

Analysis of morphological changes and the expression of epithelial-mesenchymal transition (EMT)-related factors in ovarian cancer cells after cigarette smoke extract (CSE) exposure. (A) Long-term exposure to CSE induces a mesenchymal morphology in ovarian cancer cells. Cell morphology was observed under an inverted phase-contrast microscope (Olympus, Tokyo, Japan). The magnification bar is 100 μm. The photographs were taken at a 3,100 magnification using a digital camera. (B) CaOV3 and SKOV3 cells were exposed to 5% CSE for 0, 1, 2 and 4 weeks. Total cell lysates were immunoblotted with an antibody against E-cadherin, N-cadherin, vimentin, ZO-1, α-SMA, snail or β-actin, which served as an internal control. (C) The migratory capacity and invasiveness of the ovarian cancer cells were both increased following CSE treatment, as detected by the transwell migration assay kit and the basement membrane extract (BME) cell invasion assay, which are described in the Materials and Methods section. Each value is the mean±SD of three measurements. *p<0.01. (D) The concentrations of transforming growth factor-beta 1 (TGF-β1), vascular endothelial growth factor (VEGF) and interleukin-8 (IL-8) in the culture supernatants of CaOV3 and SKOV3 cells were quantified by ELISA. The cells were seeded into 6-well plates (1.5×105/well) and treated with 5% CSE for >4 weeks. *p<0.01. The data are presented as the mean of three independent experiments and the error bars represent the SDs of the means. The results are representative of three independent experiments. RFU, Relative fluorescence units.

Cigarette smoke extract (CSE) exposure activates the p110δ-PAK1 pathway in CaOV3 cells and the p110β-ROCK1/2 pathway in SKOV3 cells. CaOV3 and SKOV3 cells were exposed to 5% CSE for >4 weeks. (A, B) Total cell lysates were immunoblotted with antibodies against (A) p110α, p110β, p110γ, p110δ, (B) p-PAK1, PAK1, p-PAK4, PAK4, ROCK1, ROCK2, p-LIMK1, LIMK1 or β-actin, which served as an internal control. (C-E) The cells were treated with 1 μM of the p110δ specific inhibitor CAL-101 or 1 μM of the p110β specific inhibitor TGX-221 for 24 h. Total cell lysates were immunoblotted with antibodies against (C) p-LIMK1, LIMK1, p-PAK1, PAK1, p110δ, ROCK1, ROCK2, (D) E-cadherin, N-cadherin or α-SMA. β-actin served as the loading control. (E) Activated RhoA was pulled-down by glutathione-S-transferase linked to the RhoA-binding domain of Rhotekin (GST-RBD) and analyzed by immunoblotting. (F) The cells were treated with 5 μM of the PAK1-specific inhibitor IPA-3 or 1 μM of the ROCK1/2-specific inhibitor RKI-1447 for 24 h. Total cell lysates were immunoblotted with antibodies against p110β, p110δ, p-PAK1, PAK1, ROCK1, ROCK2, p-LIMK1, LIMK1 or β-actin, which served as an internal control. The results are representative of three independent experiments.
TGF-β1-induced ROS generation induces PI3K-PAK1 or PI3K-ROCK1/2 signaling pathways. We next investigated whether the secreted TGF-β1 in CSE-exposed ovarian cancer cells positively regulates EMT processes and PAK1 or ROCK1/2 activation to produce EMT-related cytokines. Treatment with LY2109761 (a small-molecule inhibitor of the TGF-β receptor type I/type II) in CSE-treated ovarian cancer cells significantly blocked the expression of mesenchymal markers (Figure 4A) and reversed the fibroblast-like morphology back to the original epithelium-shaped cells (Figure 4B). The inhibition of TGF-β1-induced signaling by LY2109761 suppressed PAK1 and LIMK1 activation in CaOV3 cells and ROCK1/2 and phosphorylated LIMK1 expression in SKOV3 cells (Figure 4C). Targeted inhibition of the TGF-β1-mediated signaling pathway using LY2109761 not only effectively blocked cell motility and invasive activity but also prevented the production of EMT-related cytokines by CSE-stimulated ovarian cancer cells (Figure 4D and E). TGF-β1 contributes to tumor invasion and metastasis through the ROS-mediated activation of mitogen-activated protein kinases or nuclear factor-κB (NF-κB) (33). Furthermore, TGF-β1 induces ROS production, which, reciprocally, activates TGF-β1 production (34). We next investigated whether TGF-β1-induced ROS regulates the PAK1- or ROCK1/2-mediated LIMK1 activation to induce EMT processes. The production of ROS in CSE-exposed ovarian cancer cells was profoundly suppressed following treatment with LY2109761 and ROS scavenger, NAC (Figure 5A). Meanwhile, an inhibitor of PI3K had no effect on the level of ROS in the CSE-activated ovarian cancer cells (Figure 5A). These results suggest TGF-β1-induced ROS production initiates the PI3K-PAK1- or PI3K-ROCK1/2-mediated LIMK1 activation in CSE-exposed ovarian cancer cells. Furthermore, blocking the production of ROS by treatment with NAC prevented the activation of p110δ, the expression of phosphorylated PAK1 and LIMK1, as well as the up-regulation of N-cadherin and α-SMA in CSE-exposed CaOV3 cells (Figure 5B and C). NAC also suppressed the expression of p110β, ROCK1/2 and phosphorylated LIMK1, as well as the expression of mesenchymal markers in CSE-treated SKOV3 cells (Figure 5B and C). EMT-related cytokines, including IL-8, VEGF and TGF-β1, were efficiently down-regulated after treatment with NAC in CSE-stimulated ovarian cancer cells (Figure 5D). These results suggest that the TGF-β1 produced by CSE-treated ovarian cancer cells plays an important role in activation of PAK1, ROCK1/2 and LIMK1, leading to strengthened TGF-β1 production.
Activation of LIMK1 is a common pathway to produce TGF-β1 in CSE-exposed ovarian cancer cells. Finally, we examined how LIMK1 activation modulates morphological changes, the induction of mesenchymal markers and migratory activity in ovarian cancer cells after long-term culture in the presence of CSE. Knockdown of LIMK1 in CSE-exposed ovarian cancer cells resulted in cell morphology changing from fibroblast-like to an epithelial one (Figure 6A) and reduced the expression of mesenchymal markers (Figure 6B). Gene silencing of LIMK1 by interfering with siRNA reduced the invasion and motility of CSE-stimulated ovarian cancer cells (Figure 6C), production of TGF-β1 (Figure 6D) and generation of ROS as well (Figure 6E). However, targeted inhibition of LIMK1 had no effect on the activation of PAK1 and ROCK1/2 in CSE-treated ovarian cancer cells (Figure 6F). These results suggest that the activation of LIMK1 modulates TGF-β1-mediated EMT processes in CSE-stimulated ovarian cancer cells.
Discussion
Cancer patients who continue to smoke following their diagnosis have higher recurrence rates or metastasis of the primary tumor compared to non-smokers (35). The incidence and risk of borderline ovarian cancer increases in women who had smoked or continue to smoke compared to never smokers (36). However, the effect of cigarette smoke on ovarian cancer, especially on the primary and metastatic sites, is still unclear. In this study, long-term exposure to CSE up-regulated the mesenchymal characteristics and TGF-β1 production in both primary and metastatic ovarian cancer cells. Treatment of CaOV3 cells with CSE resulted in Src-dependent p110δ activation, which led to the activation of the PAK1/LIMK1 signaling pathway. Furthermore, Lyn-induced p110β pathway activation initiated ROCK1/2-mediated LIMK1 expression in SKOV3 cells after culture in the presence of CSE (Figure 7). These results suggest that CSE triggers different p110 isotypes to enhance the migration of ovarian cancer cells in a cell-type dependent manner and that pharmacological agents against specific p110 isoforms might be a new therapeutic modality for modulating the metastasis of ovarian cancer. CSE exposure decreases the expression of epithelial markers and concomitantly increases the expression of mesenchymal markers in lung cancer cells (37). The production of matrix metalloproteinase-1 (MMP-1), MMP-3 and MMP-10 is also increased after treatment with CSE in airway smooth muscle cells (38). Breast cancer cells acquire mesenchymal properties, obtain stem-like markers and acquire more invasive characteristics after chronic low-dose exposure to CSE (1). Rac1, a member of the Rho family, is required for fibroblast and breast cancer cell migration (39, 40). Targeted inhibition of Rac1 prevents CSE-induced Akt activation and TGF-β1 release from pulmonary epithelial cells (28). PAK1, one of downstream effectors of Rac1, has an important role in cell survival, proliferation and motility (41). Lysophosphatidic acid-stimulated PI3K/PAK1 is required for the progression and metastasis of breast cancer cells (42). Interaction with Rac1 and PAK1 is also necessary for TGF-β-induced actin rearrangement and migration of breast cancer cells (43). Because PI3K-dependent PAK or Rac1-mediated PI3K signaling pathways contribute to the metastasis of breast cancer, we anticipated that long-term stimulation with CSE activates both the PI3K/Rho/ROCK and PI3K/PAK1 signaling pathways. The PI3K/Akt signaling pathway was also activated in CSE-exposed ovarian cancer cells, whereas treatment with TGX-221 (a specific p110β inhibitor) down-regulated RhoA activity and its downstream target, ROCK1/2, in SKOV3 cells. CSE-exposed CaOV3 cells induced the expression of p110δ isoform of PI3K and PAK1, but did not alter p110β or ROCK1/2. These results suggest that p110β-ROCK1/2- or p110δ-PAK1-mediated TGF-β1 production can be a cell-type-specific target for controlling metastasis and invasion in CSE-stimulated ovarian cancer cells. These results also indicate that the selective inhibition of the p110 isoform of PI3K modulates EMT processes in primary and metastatic ovarian cancer cells after stimulation with CSE.

The Src family of kinases induces the PI3K-mediated epithelial-mesenchymal transition (EMT) process in cigarette smoke extract (CSE)-exposed ovarian cancer cells. CaOV3 and SKOV3 cells were exposed to 5% CSE for >4 weeks. (A) Total protein was extracted from cell lysates and subjected to western blotting for p-Src, Src, p-Lyn, Lyn, p-Fyn or Fyn. (B-D) The cells were treated with the dual Src and Lyn inhibitor PP1 (200 nM) for 24 h. (B) Total cell lysates were immunoblotted with the indicated antibodies. β-actin was used as the loading control. (C) The migratory capacity and invasiveness of CSE-exposed ovarian cancer cells were decreased following PP1 treatment, as detected using the transwell migration assay kit and the basement membrane extract (BME) cell invasion assay, which are described in the Materials and Methods section. Each value is the mean±SD of three measurements. *p<0.01. (D) The concentrations of transforming growth factor-beta 1 (TGF-β1), vascular endothelial growth factor (VEGF) and interleukin-8 (IL-8) in the culture supernatants of CaOV3 and SKOV3 cells were quantified by ELISA. *p<0.01. The data are presented as the mean of three independent experiments and the error bars represent the SDs of the means. RFU, Relative fluorescence units.

Transforming growth factor-beta 1 (TGF-β1) signaling promotes the PI3K-PAK1 or PI3K-ROCK1/2 signaling pathways. CaOV3 and SKOV3 cells were exposed to 5% cigarette smoke extract (CSE) for >4 weeks. The cells were treated with the dual transforming growth factor-beta (TGF-β) receptor I and II kinase inhibitor LY2109761 (100 nM) for 24 h. (A, C) Total cell lysates were immunoblotted with antibodies against (A) E-cadherin, N-cadherin, α-SMA, (C) p-LIMK1, LIMK1, p-PAK1, PAK1, ROCK1, ROCK2, p110β or p110δ. β-actin was used as the loading control. (B) Inhibition of the TGF-β1 signaling pathway following LY2109761 treatment suppressed the mesenchymal morphology in ovarian cancer cells. The morphology of cells was observed under an inverted phase-contrast microscope (Olympus, Tokyo, Japan). The magnification bar is 100 μm. Photographs were taken at a 3,100 magnification using a digital camera. (D) The migratory capacity and invasiveness of CSE-exposed ovarian cancer cells were inhibited following LY2109761 treatment, as detected by the transwell migration assay kit and the basement membrane extract (BME) cell invasion assay, which are described in the Materials and Methods section. Each value is the mean±SD of three measurements. *p<0.01. (D) The concentrations of transforming growth factor-beta 1 (TGF-β1), vascular endothelial growth factor (VEGF) and interleukin-8 (IL-8) in the culture supernatants of CaOV3 and SKOV3 cells were quantified by ELISA. *p<0.01. The data are presented as the mean of three independent experiments and the error bars represent the SDs of the means. RFU, Relative fluorescence units.

Transforming growth factor-beta 1 (TGF-β1)-induced reactive oxygen species (ROS) generation activates the PI3K-PAK1 or PI3K-ROCK1/2 signaling pathways. CaOV3 and SKOV3 cells were exposed to 5% cigarette smoke extract (CSE) for >4 weeks. (A) The cells were pretreated with 10 μM DCFA-DA for 30 min and then treated with the dual TGF-β receptor I and II kinase inhibitor LY2109761 (100 nM), the ROS scavenger NAC (10 mM), the dual Src and Lyn inhibitor PP1 (200 nM), the p110δ specific inhibitor CAL-101 (1 μM) or the p110β specific inhibitor TGX-221 (1 μM) for 24 h. The cell were transfected with LIMK1 siRNA (200 nM) or control siRNA for 48 h prior to the experiments. The cells were analyzed by flow cytometry. The numbers in the 2,7-dichloroflorescin (DCF) histograms show the mean fluorescence intensity (MFI). Thin-line histograms represent the ROS levels of the negative control cells. Thick-line histograms represent the ROS levels of the indicated drug-treated cells. (B, C) The cells were treated with the ROS scavenger NAC (10 mM) for 24 h. Total protein was subjected to Western blot analysis and immunoblotted against (B) p-LIMK1, LIMK1, p-PAK1, PAK1, ROCK1, ROCK2, p110β, p110δ, (C) E-cadherin, N-cadherin or α-SMA. β-actin was used as the loading control. (D) The concentrations of TGF-β1, vascular endothelial growth factor (VEGF) and interleukin-8 (IL-8) in the culture supernatants of CaOV3 and SKOV3 cells were quantified by ELISA. *p<0.01. The data are presented as the mean of three independent experiments and the error bars represent the SDs of the means.

Activation of LIMK1 is a common pathway to produce transforming growth factor-beta 1 (TGF-β1) in cigarette smoke extract (CSE)-exposed ovarian cancer cells. CaOV3 and SKOV3 cells were exposed with 5% CSE for >4 weeks. The cells were seeded into 6-well plates (1.5×105/well) and transfected with LIMK1 siRNA (200 nM) or control siRNA for 48 h prior to the experiments. (A) LIMK knockdown inhibited mesenchymal morphology in ovarian cancer cells. The morphology was observed under an inverted phase-contrast microscope. The magnification bar is 100 μm. The photographs were taken at a 3,100 magnification using a digital camera. (B) Total protein was subjected to Western blot analysis and immunoblotted against E-cadherin, N-cadherin or α-SMA. (C) The migratory capacity and invasiveness of CSE-exposed ovarian cancer cells were inhibited by LY2109761 treatment, as detected by the transwell migration assay kit and the basement membrane extract (BME) cell invasion assay, which are described in the Materials and Methods section. Each value is the mean±SD of three measurements. *p<0.01. (D) The concentrations of TGF-β1 in the culture supernatants of CaOV3 and SKOV3 cells were quantified by ELISA. *p<0.01. The data are presented as the mean of three independent experiments and the error bars represent the SDs of the means. (E) Total protein was subjected to western blot analysis and immunoblotted against p-LIMK1, LIMK1, p-PAK1, PAK1, ROCK1, ROCK2, p110β or p110δ. β-actin was used as the loading control. The results are representative of three independent experiments.

Proposed model depicting the signaling pathway of cigarette smoke extract (CSE)-induced epithelial-mesenchymal transition (EMT) in human ovarian cancer cells. CSE exposure induces the secretion of active transforming growth factor-beta 1 (TGF-β1) in ovarian cancer cells. Specifically, CSE exposure increased the activity of Src, p110δ and PAK1 in CaOV3 cells and the activity of Lyn, p110β and ROCK1/2 in SKOV3 cells. LIMK1 is a common pathway of TGF-β1 in CSE-exposed ovarian cancer cells. The LIMK1 pathway can activate TGF-β1. These signaling molecules change EMT markers, namely by increasing the expression of N-cadherin, vimentin, Snail and α-SMA and decreasing the expression of E-cadherin and ZO-1.
PAK- or ROCK-mediated LIMK1 activation is closely related to cell migration and invasion (44, 45). Increased LIMK1 phosphorylation in breast cancer, prostate cancer and gastric cancer is highly connected with the invasiveness of the cancer cells (24, 46). The overexpression of LIMK1 leads to an elevated collagenolytic activity of MMP-2 and secretion of pro-MMP2 and pro-MMP-9 (46). Furthermore, the levels of phosphorylated PAK1 and LIMK1, one of the downstream targets of PAK1, were higher in migratory and invasive lung cancer cells (47). Meanwhile, osteopontin triggers PI3K/AKT-mediated ROCK1 expression, but not PAK1/2, to induce the invasion of lung cancer cells (48). By contrast, high Rho activity repressed p21 expression to inhibit cell growth and prolonged expression of LIMK1 restored stress fibers and reduced motility (49). Therefore, it is likely that PAK1 or ROCK1/2 expression alone is not involved in LIMK1 expression to determine the motility and invasion status of carcinoma cells. Because of the differences in p110 isoform activity in CSE-exposed ovarian cancer cells, we observed that the targeted inhibition of PAK1 in CaOV3 cells or ROCK in SKOV3 cells effectively reduced LIMK1 phosphorylation and activation after CSE stimulation in this study. In addition, gene silencing of LIMK1 with siRNA significantly inhibited the invasive activity and secretion of EMT-related cytokines in both CSE-activated primary (CaOV3) and metastatic (SKOV3) ovarian cancer cells. These results suggest that the TGF-β1-LIMK1 activation pathways are not identical but LIMK could be a common attractive therapeutic target in CSE-derived TGF-β1 release and metastasis of ovarian cancer.
ROS initiate diverse downstream signal transduction cascades through TGF-β1, which is related to tumorigenesis and cell motility (33, 34). Moreover, increased ROS levels may, in turn, increase TGF-β1 expression and stimulate the release of TGF-β1 (50). Treatment with LY2109761 or NAC in CSE-exposed ovarian cancer cells reduced ROS generation and resulted in TGF-β1 secretion. Meanwhile, targeted inhibition of Src kinase, PI3K and LIMK failed to block ROS secretion. These results suggest that TGF-β1-mediated ROS triggers the PI3K-PAK1-LIMK1 or PI3K-ROCK1/2-LIMK1 pathways and strengthens TGF-β1 production.
In conclusion, we first report that CSE stimulation dominantly activates the p110δ-PAK1 signaling pathway in primary ovarian cancer and the p110β-ROCK1/2 pathway in metastatic ovarian cancer. Our results suggest that cigarette smoke-induced activation of a particular p110 isoform could be a new pathophysiological concept and therapeutic target to control migration and invasion of ovarian cancer. Thus, this result supports the idea that identifying the specific p110 isoform is necessary for clinically controlling ovarian cancer.
Acknowledgements
This study was supported by the Basic Science Research Program of Ministry of Education (NRF-2015R1D1A1A01056672) and Ministry of Science, ICT & Future Planning (NRF-2015R1C1A2A01053732) through the National Research Foundation (NRF) of the Republic of Korea.
Footnotes
Conflicts of interest
The Authors declare that they have no conflicts of interest to disclose.
- Received January 31, 2017.
- Revision received March 5, 2017.
- Accepted March 6, 2017.
- Copyright© 2017, International Institute of Anticancer Research (Dr. George J. Delinasios), All rights reserved





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}