


Abstract
Background/Aim: One of the major hurdles in the treatment of breast cancers is the inability of anti-cancer drugs to eliminate the breast cancer stem cells (BCSCs) population, which leads to disease relapse. The dearth in anti-cancer drugs that target BCSCs can be attributed to the absence of in vitro screening models that can not only recapitulate the tumor microenvironment consisting of BCSCs but also preserve the 3-dimensional (3D) architecture of in vivo tumors. Materials and Methods: In our present study, we have developed a 3D cell culture system that shows: (i) enrichment of BCSCs, (ii) increased drug resistance, and (iii) generation of hypoxic conditions similar to tumors. Results: Using this model, we were able to screen a FDA-approved diversity set and identify as well as validate actinomycin D as a potential anti-breast cancer agent. Interestingly, we show that actinomycin D specifically targets and down-regulates the expression of the stem cell transcription factor, Sox-2. Additionally, down-regulation of Sox-2 leads to depletion of the stem-cell population resulting in the inability of breast cancer cells to initiate tumor progression. Conclusion: This study demonstrates the utility of an in vivo-like 3D cell culture system for the identification and validation of anti-cancer agents that will have a better probability of success in the clinic.
Breast cancer, although generally referred to as a single disease, consists of at least 21 distinct histological subtypes and up to 4 different molecular subtypes (1). It is estimated that 231,840 cases were diagnosed in a year and approximately 40,290 are expected to die of the disease (www.cancer.org). It is not surprising that heterogeneity in the types of breast cancer plays a huge role in determining its severity and treatment strategy. However, one common facet that holds true across tumor types is the inability of available anti-breast cancer therapy to eliminate and cure the disease.
One of the driving forces that leads to relapse in breast cancer after treatment is breast cancer stem cells (BCSCs). BCSCs have been shown to be highly tumorigenic and extremely resistant to therapy (2, 3). Even though there is a large disparity in characterization of BCSCs, they all show ability to differentiate, have the capacity of tumor initiation and generally show up-regulation of one of the many stem cell transcriptional factors (4). Interestingly, the CD44highCD24low subset of the breast cancer cell population has been shown to have all the above stated properties and is a good biomarker for identification of stem cells (5). Even with this plethora of knowledge it has been hard to target BCSCs because of their low abundance in tumors and a complete absence of an in vitro model that sustains the population of these cells. The most common method is to utilize sphere forming assays to screen for drugs that inhibit tumorsphere formation (6). Unfortunately, these types of assays identify drugs that inhibit the bulk of the proliferating cells and thus are not beneficial in the long run.
Another hurdle in the path to identification of a cure for breast cancer is the huge cost associated in the process of new drug discovery (7). One way to circumvent this is to utilize agents that are already in clinical use for a variety of cancer treatments. The impetus for repurposing FDA-approved agents is derived from the fact that drug development is lengthy and expensive and that these agents have already passed toxicology assessments to demonstrate safety (8). In light of this, our hypothesis is that the use of a cell culture system that faithfully mimics in vivo tumors (taking into consideration BCSCs) to screen for clinically approved anti-cancer drugs will yield hits that will ensure efficacy in clinical trials in an accelerated time and at a lower cost.
To address this, we had previously developed a novel fiber-inspired smart scaffold (FiSS) that allowed the growth of cancer cells in the form of a organoid called tumoroid (9). Tumoroids were characterized and demonstrated to mimic in vivo tumors. In the present study we utilized the above platform in combination with an FDA-approved NCI oncology diversity set in order to identify and validate a clinically approved drug for targeting and eliminating BCSCs. During this process we have shown that actinomycin D is a very potent compound that specifically targets and down-regulates stem cell transcription factor SRY (sex determining region Y)-Box-2 (SOX2), leading to stem cell depletion within the tumor bulk.
Materials and Methods
Cell cultures. MCF-7, MDA-MB-231 and BT474 were purchased from ATCC. Doxorubicin resistant MCF-7 (MCF-7/dox) was obtained from Dr. Robert A. Gatenby (Moffitt Cancer Center, Tampa, FL, USA) and maintained in growth media containing 1 μM of doxorubicin. All cell lines were maintained in RPMI-1640 supplemented with 10% fetal Bovine serum and 1X penicillin/streptomycin in an incubator at 37°C with 5% CO2.
Synthesis of cobalt chloride (CoCl2) scaffold. For the synthesis of CoCl2 scaffold, 2.4 g of PLGA, 0.6g of mPEG/PLA, and 0.195 mg of CoCl2, dissolved in methanol (5 mg/ml) were dissolved in 10 ml of 80/20 dichloromethane/chloroform and the mixture was electrospun, as described previously (9).
Tumoroid culture and flow cytometry. MCF-7 cells were seeded onto FiSS (Transgenex nanobiotech, Tampa, FL, USA) in 6-well plates at 240,000 cells per well. Cells were cultured for six days in RPMI media to form primary tumoroids. Primary tumoroids were dissociated by treatment with trypsin and collected for seeding of secondary scaffolds. Cells from primary tumoroids were seeded onto secondary scaffolds in 6-well plates at 240,000 cells per well. These scaffolds were also cultured for six days to generate secondary tumoroids. Media was changed on the fourth day and media containing either vehicle control, 1.56 nM, or 6.25 nM actinomycin D was added.
Scaffolds were treated with 1 mL accutase per well for 8 min at room temperature on an orbital shaker to dissociate the tumoroids. Scaffolds were washed with PBS to collect cells for analysis by flow cytometry. Secondary tumoroid cells were resuspended in cold FACS buffer (PBS containing 10% FBS and 2.5 mM EDTA) and stained using fluorochrome conjugated antibodies for CD24 (Becton Dickenson, Franklin Lakes, NJ, USA) and CD44 (Miltenyi Biotec Inc., San Diego, CA, USA). Isotype control antibodies were used to identify any non-specific binding. Compensation for spectral overlap was performed and data was collected using a FACSCanto II flow cytometer (Becton Dickenson, Franklin Lakes, NJ, USA).
Measurement of IC50 of the drugs. FDA-approved Oncology Diversity set was obtained from NCI in 96 well plate format with each well containing 20 μl of 10 mM drug stock. All different cell lines were cultured either in regular 96-well plate without scaffold (served as monolayer) or with FiSS (scaffold culture) in complete medium. For scaffold culture, 75% medium was carefully removed and replaced with 150 μl of fresh medium every alternate day. After five/six days of scaffold culture, cells were stained with NucBlue (Thermo Fisher Scientific, Waltham, MA, USA) to check if tumoroids had formed and drug was added in triplicates with varying concentrations. The cells were incubated for 48 h in a humidified atmosphere under 5% CO2 at 37°C then cell viability was measured using Presto-blue (Fisher Scientific) through measure of the absorbance at 570 nm. The IC50 was calculated from the dose response curve using GraphPad Prism Software version 5.01 (GraphPad Software Inc., La Jolla, CA, USA).
Real time polymerase chain reaction. MCF-7 and MCF-7/dox cells were seeded as monolayer and 3D tumoroid cultures (both in controls and CoCl2 scaffold cultures) in 96 well plates. Actinomycin D was treated at the concentration based on IC50 for 24 h. Accutase was used to detach the cells from the plate/scaffold platform. Cell pellet was dissolved in Trizol and RNA was isolated. RT-PCR was performed using a 2 step process namely: 1) cDNA synthesis using DNAase-1 treated RNA, and 2) PCR using primers of the gene of interest.
Detection of hypoxia. Hypoxic region of the 3D scaffold cultures was detected using Hypoxia detection kit (Enzo Life Sciences, Farmingdale, NY, USA), which was designed for functional detection of hypoxia in live cells using fluorescent microscopy. This kit includes fluorogenic probes for hypoxia (red), which take advantage of the nitroreductase activity present in hypoxic cells by converting the nitro group to hydroxylamine (NHOH) and amino (NH2) and releasing the fluorescent probe. Briefly, day six scaffold culture was incubated with the hypoxia dye for 30 min and incubated at 37°C followed by PBS washing for two times. Red fluorescence was detected using the fluorescence microscope.
Western blotting. MCF-7 and MCF-7/dox cells were seeded as monolayer and 3D tumoroid cultures in 96-well plates. Actinomycin D was used in treatments at a concentration of 50 nM based on IC50 for 24 h. Accutase was used to detach the cells from the plate/scaffold platform. Cell pellet was re-suspended in RIPA buffer and vortexed for 30 min followed by centrifugation at 4°C at 13000 rpm to collect the protein extract. Protein content was determined using the BCA method followed by western blot procedure. Antibodies for NANOG, OCT4A, SOX2 and HPRT were obtained from Cell Signaling Technology Inc. (Danvers, MA, USA).
Statistical analysis. All experiments were performed three independent times and the data was represented as mean±standard error of mean (SEM), unless otherwise noted in the legend. Statistical comparison between treatment groups was made using one-way ANOVA followed by post-hoc Tukey's test. A p<0.05 was considered as statistically significant.
Results
Increase in BCSCs within the tumoroids correlates with increased drug resistance. We have utilized FiSS, which has been shown to induce growth of 3D tumoroids that mimic in vivo tumors (9). For this study, we utilized MCF-7 and its syngeneic drug resistant cell line MCF-7/dox (10). We hypothesized that MCF-7 tumoroids and MCF-7/dox tumoroids should show induction of BCSCs, which in turn should lead to increased resistance irrespective of their intrinsic drug sensitivity. MCF-7 and MCF-7/dox formed well-defined tumoroids (Figure 1A) and a doxorubicin dose response curve confirmed that MCF-7/dox (IC50: 4.4±0.28 μM) was 24-fold more resistant than MCF-7 (IC50: 0.18±0.008 μM) in monolayer (Figure 1B). Not surprisingly, doxorubicin showed higher IC50 in the tumoroids of MCF-7/dox (1.31±0.27 μM in MCF-7 vs. 23.09±5.35 μM in MCF-7/dox) while maintaining the differences in drug sensitivity. Dose escalation studies using doxorubicin in humans have shown that the highest plasma concentration reached is about 383 ng/ml, which is in the range of the IC50 of MCF-7 tumoroids (11). Therefore, the 3D cell culture platform in this study could be an efficient platform for screening of drugs for potential anti-cancer activity.

Tumoroids show increased drug resistance compared to cells grown in monolayer. (A) MCF-7 and MCF-7/dox cells were cultured on the scaffold for six days and then visualized using Nuc-blue. (B) MCF-7 and MCF-7/dox cells were cultured on scaffold for six days and then cells were treated with increasing concentration of doxorubicin. Forty-eight hours post-treatment cell viability was analyzed by measuring absorbance value at 570 nm using presto-blue as per manufacturer's instructions. From the relative absorbance value and used drug concentrations the IC50 value was calculated using GraphPad Prism. Data are represented as Mean±SEM of three independent experiments performed in triplicates (*denotes p<0.05).
Since the presence of BCSCs, as defined by the CD44highCD24low cell population, positively correlates with shorter overall survival in patients, it is imperative to test potential drugs on a platform that amplifies and maintains BSCSs (12). As seen in Figure 2A, MCF-7 grew into well-formed single cell tumoroids after six days in culture. More importantly, these tumoroids showed a 3-fold amplification of CD44highCD24low cells when compared to monolayer cells (Figure 2B). To further confirm that the growing tumoroids harbored BCSCs, we looked at the transcript and the protein levels of the stem cell transcription factors OCT4A, SOX2 and NANOG (13). On comparing the gene expression profile using real-time RT-PCR, we found that irrespective of the basal monolayer expression, transcript levels of all the transcription factors were increased in the tumoroids compared to the cells growing as a monolayer, in both MCF-7 and MCF-7/dox cells. Specifically, NANOG, OCT4A and CXCR4 levels were all statistically higher in MCF-7 cells grown on scaffold. However, only OCT4A levels were statistically significant (p<0.05) in MCF-7/dox cells grown on scaffold (Figure 2C). Furthermore, as seen in Figure 2D, the transcription factors NANOG and OCT4A protein expression were higher in the MCF-7/dox monolayers compared to the MCF-7 cells, and at the same time SOX2 expression was lower in MCF-7/dox monolayer compared to the MCF-7 cells. Interestingly, out of the three, only SOX2 protein levels was increased in tumoroids compared to monolayer cells. Taken together, these results show that tumoroids amplify and maintain BCSCs and should provide a clinically relevant platform for screening drugs that target cancer stem cells.
Tumoroids faithfully recapitulate hypoxia when exposed to CoCl2. The inability of current therapy to eradicate BCSCs has been demonstrated to be the major reason for its failure in treating breast cancers (14). Since we have shown that tumoroids induce a BCSC population, we investigated further whether stem cell enrichment in tumoroids could associate with drug resistance. Hypoxia has been shown to increase drug resistance by inducing a switch to a stem-like phenotype in breast cancer cells (15). Since CoCl2 has been successfully used to mimic hypoxia in monolayer cultures of breast cancer cells, we were interested in observing the effects of CoCl2 in our 3D cell culture platform (16). For this study, we cultured MCF-7 and MCF-7/dox cells in control (normoxia) vs. CoCl2 doped FiSS (hypoxia). We allowed tumoroids to form and observed that the MCF-7/dox tumoroids (121.7±15.2 μM) were at least twice as big in size compared to MCF-7 tumoroids (67.8±6.34 μM) in normoxia and statistically significant (p<0.05) (Figure 3A). Interestingly, this difference in size was completely abolished when tumoroids were allowed to form in the presence of CoCl2 (91.3±3.02 μM in MCF-7 vs. 77.7±5.6 μM in MCF-7/dox). Next, in order to confirm that treatment with CoCl2 did indeed lead to depletion of oxygen content within the tumoroids, we utilized a red hypoxic dye as detailed in the methods section. We found that MCF-7/dox tumoroids have an intrinsic hypoxic phenotype compared to the wild-type MCF-7 tumoroids (Figure 3B). Exposure to CoCl2 led to further enhancement of hypoxia in both MCF-7 and MCF-7/dox tumoroids, with the MCF-7/dox tumoroids showing the higher intensity of red fluorescence. Furthermore, since hypoxia has been shown to lead to a glycolytic switch in cancer cells, we looked at lactic acid accumulation in the growth media in normoxia and hypoxia (17). We have shown that in accordance with tumoroid size, MCF-7/dox tumoroids showed higher amounts of lactate release and exposure to CoCl2 lead to an equivalent increase in lactate in both the MCF-7 and the MCF-7/dox tumoroids (data not shown).

Stem cell characterization of cells growing in monolayer versus tumoroids. (A) MCF-7 cells were plated on the scaffold for six days and the resulting first-generation tumoroids were visualized using Nuc-blue. (B) Single-cell suspension of the MCF-7 tumoroids were stained with APC-Cy7 conjugated CD44 antibody and APC conjugated CD24 antibody and the CD44highCD24low cells were detected using flow cytometry and analyzed using FlowJo software. (C-D) MCF-7 and MCF-7/dox cells were grown as a monolayer in cell culture dish or as a tumoroid on the scaffold. After six days in culture, the monolayer and the tumoroids were harvested and processed for RNA and protein extraction. The extracted RNA was subjected to qRT-PCR and the extracted protein was subjected to western blotting. HPRT transcript was used for normalization of qRT-PCR data and HPRT protein was used as loading control for the western blot. Data are represented as Mean±SEM of three independent experiments (* denotes p<0.05).
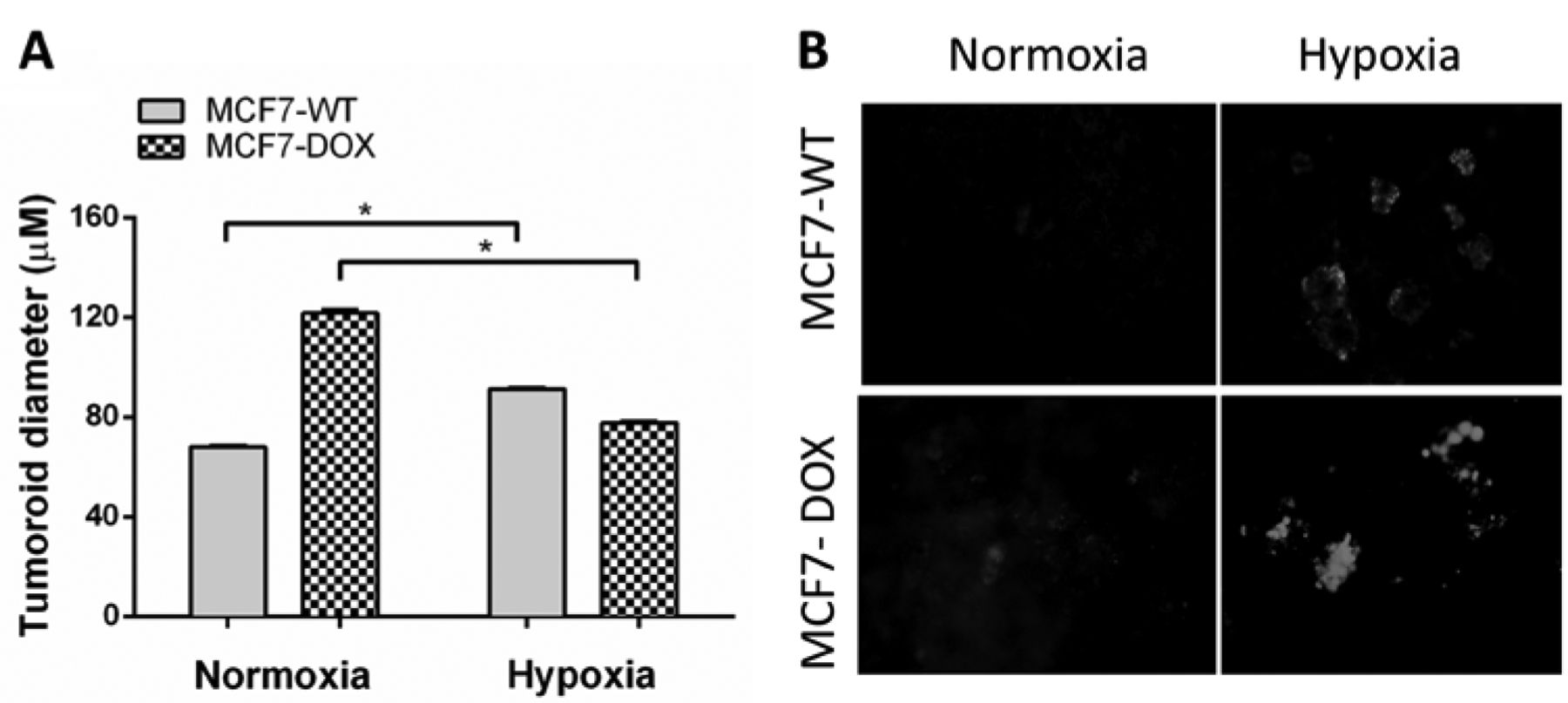
Cobalt chloride infused scaffold induces hypoxia in tumoroids. (A) MCF-7 and MCF-7/dox cells were cultured on the scaffold containing 50 μM CoCl2 for six days and then visualized using Nuc-blue. Images were captured using a fluorescence microscope and the diameter of the tumoroids were analyzed using ImageJ. (B) Six day old tumoroids grown on CoCl2 infused scaffold were stained with hypoxic dye as per the manufacturers. Data are represented as Mean±SEM of three independent experiments performed in triplicates (*denotes p<0.05).
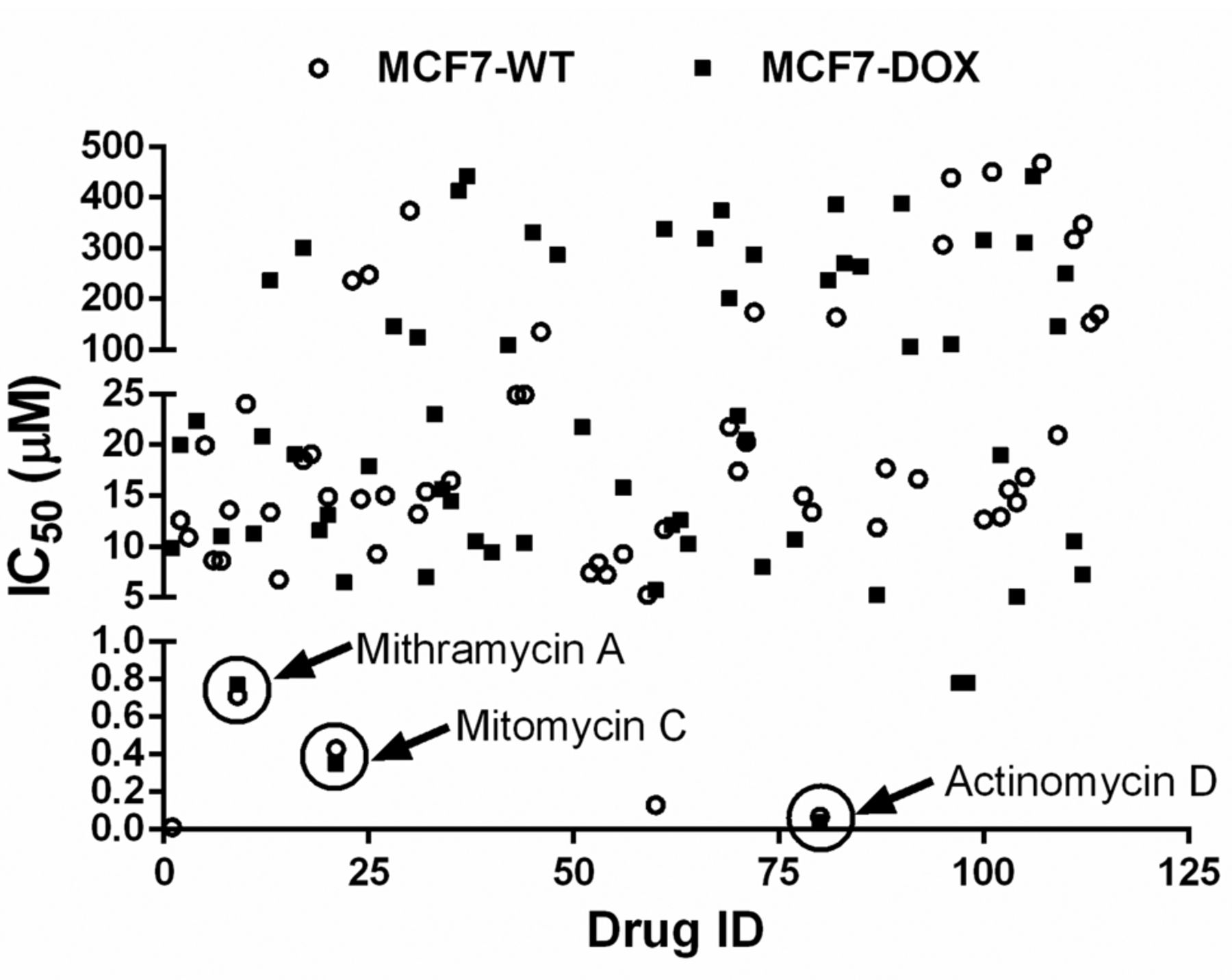
High throughput screening for compounds inducing cell death in breast cancer. In earlier studies we had characterized the 3D cell culture platform and demonstrated that it mimics all the features of in vivo tumors. Towards utilizing this platform for drug discovery, we first used MCF-7 and MCF-7/dox in a monolayer in a 96-well plate and treated them with a NCI Oncology diversity set containing FDA-approved anti-cancer drugs (https://dtp.cancer.gov). Each drug was used in a 4 point log-dose from 0.1 to 100 μM. Untreated cells were used as control and the cell death was estimated at the end of 48 h post-treatment. Figure 4 shows the IC50 values of all the drugs tested in MCF-7 and MCF-7/dox cell lines. Actinomycin D, mithramycin A and mitomycin C all showed high potency (low IC50) in both MCF-7 and MCF-7/dox cells. We validated our three hits in a panel of breast cancer cells including MCF-7, MCF-7/dox, MDA-MD-231 and BT474 cells and showed that all three molecules induced cell death in nanomolar concentration (Table I).

High throughput screening of breast cancer cells in monolayer. (A) MCF-7 and (B) MCF-7/dox cells were cultured on monolayer and then treated with increasing concentration of compounds from NCI Approved Oncology Drugs Set VII. Forty eight hours post-treatment cell viability was analyzed using presto-blue as per manufacturer's instructions. From the relative absorbance value and used drug concentrations the IC50 value was calculated using GraphPad Prism. Data are represented as Mean of experiments performed in triplicates. Black circle dot was used to indicate MCF-7 cell line and black square dot was used to indicate MCF-7/dox cell line.
Identification and validation of actinomycin D and other potential drugs using monoculture in different breast cancer cell lines.
Next, we looked at their activity on our 3D cell culture platform. Among the above three, actinomycin D showed the highest potency in inducing death in tumoroids growing in normoxia or hypoxia. Additionally, actinomycin D, unlike doxorubicin, had a very minimal increase in IC50 when MCF-7 cells in monolayer were compared to MCF-7/dox monolayer cells (0.07±0.006 μM vs. 0.037±0.021 μM in MCF-7 and MCF-7/dox, respectively) (Figure 5). Additionally, unlike mitomycin C or mithramycin A, actinomycin D was equally effective in inducing cell death in tumoroids grown in normoxic conditions and hypoxic conditions (0.78±0.25 μM vs. 1.04±0.07 μM in MCF-7 and 1.66±0.06 μM vs. 2.24±0.46 μM in MCF-7/dox, respectively) and statistically significant (p<0.05) (Figure 5). In light of this, going forward we decided to look at the mechanism involved in the actinomycin D-mediated cell death in tumoroids.
Actinomycin D depletes BCSCs by down-regulating the expression of SOX-2. In our earlier studies, we had shown that tumoroids show an increase in mRNA and protein levels of SOX2. SOX2 is an important stem cell transcription factor that has been shown to enhance tumor progression in breast cancer cells (18). Thus, we looked at the effects of actinomycin D on SOX2 protein levels in both MCF-7 and MCF-7/dox cell lines. As seen in Figure 6A, tumoroids treated with actinomycin D showed a statistically significant decrease in SOX2 protein expression in both MCF-7 and MCF-7/dox tumoroids (*p<0.05, when compared to monolayer). Interestingly, the decrease in SOX2 expression was 2-fold in both the cell lines tested when compared to the control untreated tumoroids (2.05 vs. 0.75 pixel density in MCF-7 and 1.05 vs. 0.5 pixel density in MCF-7/dox) (Figure 6B). Next, we wanted to see if the decrease in SOX2 expression would lead to decrease in the CD44highCD24low stem-like population. We found that exposure of actinomycin D to MCF-7 tumoroids decreased cell viability with IC50 29 nM (Figure 6C) and concurrently decreased the CD44highCD24low cell population as compared to untreated controls (Figure 6D). Taken together these results show that actinomycin D targets and down-regulates the expression of SOX2 resulting in depletion of the stem-like cell population, which hampers the breast cancer cell's ability to initiate tumors.

Actinomycin D is effective in inducing cell death in tumoroids. MCF-7 and MCF-7/dox cells were cultured on monolayer or on scaffold in normoxia or hypoxia for six days. At the end of culture period, the monolayer cells and tumoroids were treated with increasing concentration of actinomycin D. Forty-eight hours post-treatment cell viability was analyzed using presto-blue as per manufacturer's instructions. From the relative absorbance value and used drug concentrations the IC50 value was calculated using GraphPad Prism. Data are represented as Mean±SEM of three independent experiments performed in triplicates (*denotes p<0.05) compared to monolayer.
Discussion
The factors impeding the discovery of an effective treatment option for breast cancer include: (i) the resilience of BCSCs to avoid death in presence of treatment, (ii) the unavailability of a screening platform to identify drugs that target BCSCs, and (iii) the high cost of discovering and developing a new drug that would show promise in clinical trials. To overcome this, we have characterized a 3D cell culture system that mimics in vivo tumors. Specifically, the in vitro system presented herein allows growth of tumoroids that not only harbor and maintain BCSCs but also effectively recapitulate the hypoxic and glycolytic microenvironment of breast cancers. More importantly, we were able to demonstrate the drug resistant phenotype of tumoroids, making them a valuable platform in screening of drugs to identify effective anti-cancer agents. As proof-of-principle, using our system we have developed a screening protocol which resulted in the successful identification of the clinically approved actinomycin D as a potent anti-BCSC agent. Furthermore, we were able show that the BCSC-targeting activity of actinomycin D was based on its ability to down-regulate the protein levels of the stem cell transcription factor SOX2.

Actinomycin D treatment reduced cell viability and the CD44highCD24low sub-population in MCF-7 by downregulating SOX2. (A) MCF-7 and MCF-7/dox were allowed to form tumoroids on scaffold for 6 days, after which they were exposed to actinomycin D for 24 h. At the end of 24 h the tumoroids were dissociated and processed for western blotting. (B) Quantification of band intensity normalized to HPRT is shown. (C) Second generation MCF-7 tumoroids were cultured in a 96-well plate for six days. Tumoroids were treated with increasing concentrations of actinomycin D on day four in groups of four wells. After 48hr treatment, cell viability was detected on day six. Viability was calculated as a percent of control untreated wells and graphed using GraphPad Prism software. (D) Tumoroids were treated with actinomycin D for 48 h. Following treatment, tumoroids were dissociated and cells were co-stained using FITC conjugated CD44 antibody and Alexa Fluor-647 conjugated CD24 antibody. CD44highCD24low stem-like populations in MCF7 tumoroids were assessed by a BD FACS Canto 2 flow cytometer.
Breast cancers have been successfully cultured in 3D systems and demonstrated to more closely mimic the in vivo microenvironment and show drug resistant properties (19, 20). However, a system that recapitulates all the essential characteristics of breast cancer is lacking and at present, in vivo tumor models are still the gold standard for testing drug efficacy (21). Unfortunately, the cost and time considerations of using in vivo models prohibit their use for screening and identification of novel therapies. We had previously developed a fiber-based scaffold that allows the growth of tumor cells as a cell cluster called tumoroids that manifest markers of EMT and drug resistance (9). In our present study, we show that tumoroids contain an enrichment of BCSCs which can be maintained by induction of glycolysis and hypoxic conditions. Importantly, our system is conducive for use with high throughput screening assay platforms and allows the detection of agents that will have a high rate of success in the clinic.
One of the salient findings of this study is the identification of an FDA-approved drug, actinomycin D, and its repurposing for use in elimination of BCSCs. Actinomycin D is presently approved for use in Wilm's tumor, rhabdomyosarcoma, Ewing's sarcoma and gestational trophoblastic neoplasia (22-24). While its main mechanism of action is through inhibition of transcription (25), it has also been shown to be useful in p53-based cyclotherapy in combination regimens with anti-mitotic agents (26). Surprisingly, its effect on cancer stem cells has never been studied and thus our finding opens up an avenue for actinomycin D and its derivatives to be tested for their efficacy in different cancer models. Additionally, one of the major advantages of repurposing actinomycin D as an anti-breast cancer agent is the already available pharmacokinetic, pharmacodynamics and toxicity profile of the drug, which will expedite its use for testing in clinical trials (27).
Many of the studies involving the targeting of cancer stem cells have purported SOX2 to be a druggable target for therapy because of the high incidence of SOX2 gene amplification (28). In breast cancer, even though there is no evidence of SOX2 gene amplification, SOX2 immunoreactivity has been correlated with worse clinical outcomes (29). Specifically, SOX2 has been shown to facilitate breast cancer progression by partnering with β-catenin in activating the transcription of cell cycle regulatory proteins like CCND1 (30, 31). Furthermore, SOX2 has been shown to induce breast cancer metastasis by promoting EMT through its regulation of the WNT/β-catenin pathway (32). More importantly, SOX2, via its transcriptional activation of the mTOR pathway, has been shown to be very critical in maintaining the stem cell like phenotype in breast cancer cells (33). Also, SOX2 expression correlated with development of drug resistance to standard breast cancer therapy like tamoxifen (34). Not surprisingly, targeted knock-down or epigenetic silencing of SOX2 has been shown to decrease breast cancer cell proliferation and breast cancer progression, thereby demonstrating the usefulness of SOX2-based targeted therapy (35, 36). A specific small molecule that targets SOX2 has yet to be developed.
In the present study we have demonstrated the ability of actinomycin D to down-regulate SOX2 and thereby eradicate the BCSC population in our tumoroids. This decrease has been demonstrated to occur via the suppression of SOX2 transcription leading to decrease in SOX2 protein expression. The observation that the protein expression of other stem cell transcription factors remains unchanged leads us to believe that this represents a specific targeted actinomycin D activity. One possible explanation for this specificity would be the proclivity of actinomycin D to intercalate at the GC rich regions of the SOX2 promoter (37). However, a thorough ChiP analysis of the SOX2 promoter region in actinomycin D treated and untreated cells will have to be carried out and is beyond the scope of the present study.
Taken together, our results show the usefulness of our developed 3D cell culture model to screen and identify novel anti-cancer stem cell agents. Our method to screen clinically approved drugs with the intention of finding new purposes for these drugs will help expedite drug approval while keeping the cost of drug discovery very low. Further studies regarding the packaging and delivery methods of actinomycin D (nanoparticles, antibody-targeted therapy) will need to be initiated to improve the PK/PD profile of actinomycin D for use in the clinic.
Acknowledgements
These studies were supported by SBIR contracts (Award # HHSN261201300044C, # HHSN261201400022C, HHSN261201 500065C) from NCI. Ryan Green is supported by an American Heart Association pre-doctoral fellowship. Subhra Mohapatra (founder and scientific advisor), Shyam Mohapatra (founder and scientific advisor), and the University of South Florida have equity interest in TGN. The terms of this arrangement have been reviewed and approved by the University of South Florida, Tampa, in accordance with its conflict of interest policies.
Footnotes
↵* These Authors contributed equally to this work.
- Received January 12, 2017.
- Revision received March 9, 2017.
- Accepted March 13, 2017.
- Copyright© 2017, International Institute of Anticancer Research (Dr. George J. Delinasios), All rights reserved
References
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.