


Abstract
Background: Itraconazole is a common antifungal agent that has demonstrated anticancer activity in preclinical and clinical studies. This study investigated whether itraconazole exerts this effect in endometrial cancer (EC) cells. Materials and Methods: Cell viability was evaluated with the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay, and gene and protein expression were assessed by microarray analysis and immunoblotting, respectively, in five EC cell lines. Results: Itraconazole-suppressed proliferation of AN3-CA, HEC-1A and Ishikawa cells (p<0.05) but not of HEC-50B or SNG-II cells. Itraconazole did not suppress GLI1 or GLI2 transcription but did inhibit the expression of mammalian target of rapamycin (mTOR) signaling components in AN3-CA and HEC-1A cells, while inducing that of microtubule-associated protein 1A/1B-light chain 3-II, a marker of autophagy. ATP-binding cassette transporter A1 gene was down-regulated in Ishikawa, HEC-50B and SNG-II cells. Conclusion: Itraconazole treatment suppresses the growth of EC cells by inhibiting AKT/mTOR signalling.
Endometrial cancer (EC) is the most common gynaecological malignancy in developed countries (1). Although the outcome of women with early-stage and low-grade EC is generally favourable, it remains poor in patients with advanced or recurrent disease (2). Grade 3 endometrioid and non-endometrioid EC can progress even when diagnosed at an early stage. There is no standard of care beyond the first-line treatment of six cycles of carboplatin and paclitaxel (3). As such, there is a need for the development of new treatments.
Itraconazole is a commonly used oral antifungal agent that functions by blocking ergosterol synthesis in fungal cell membrane. Itraconazole has been repositioned as an anticancer agent in both pre-clinical and clinical studies, and was found to reverse P-glycoprotein-mediated chemoresistance of cancer cells (4, 5) and inhibit angiogenesis (6) as well as Hedgehog (7, 8) and AKT/mammalian target of rapamycin (mTOR) (9, 10) signaling, which are associated with dysregulation of intracellular cholesterol transport (9, 11). Recent studies have also reported the induction of autophagy and inhibition of lymphangiogenesis by itraconazole (11, 12).
We have been treating patients with refractory solid tumours by combination itraconazole chemotherapy since 2008. A retrospective multi-institutional study demonstrated that overall survival was prolonged in patients with refractory ovarian cancer treated with itraconazole (hazard ratio=0.27, p=0.006) (13). Our review of itraconazole-treated patients showed a survival advantage among those with triple-negative breast, pancreatic, or biliary tract cancer relative to controls (14-16). In a randomized phase 2 clinical trial, combined treatment with itraconazole and pemetrexed prolonged overall survival (hazard ratio=0.194, p=0.012) in patients with non-small cell lung cancer (17).
Hedgehog signaling inhibitors have been shown to suppress proliferation of EC cells (18), and mTOR inhibitors have also been investigated for their therapeutic potential in EC (3). We previously reported itraconazole administered at a physiological dose showed dose- and time-dependent suppression of Ishikawa and HEC-1A human EC cell proliferation (19). The present study investigated whether itraconazole has the same anticancer effects in EC as in other types of cancer using five EC cell lines.
Materials and Methods
Cell culture. The human EC cell lines HEC-1A and AN3-CA were obtained from the American Type Culture Collection (Manassas, VA, USA); HEC-50B and SNG-II were obtained from the Japanese Collection of Research Biosources Cell Bank (Tokyo, Japan). Cells were cultured according to the suppliers' recommendations. Ishikawa cells were provided by Dr. Masato Nishida (Tsukuba University, Ibaraki, Japan) (18, 19). Ishikawa cells were cultured in Roswell Park Memorial Institute-1640 medium (Gibco, Grand Island, NY, USA) supplemented with 10% heat-inactivated foetal calf serum (Omega, Tarzana, CA, USA).
Cell viability assay. Cells (5×103/well) were seeded in 96-well culture plates and allowed to adhere overnight. Attached cells were treated with up to 10 μM itraconazole (Sigma-Aldrich, Tokyo, Japan) for 48 h. Cell viability was then evaluated with 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) Cell Proliferation Assay kit (Cayman Chemical Company, Ann Arbor, MI, USA) according to the manufacturer's protocol. Briefly, cells were incubated with MTT reagent for an additional 4 h, and the dissolved formazan product was measured by reading the absorbance of samples at 570 nm on a microplate reader. The experiment was repeated at least three times.
Microarray analysis of gene expression. Cells were cultured with 10 μM of itraconazole or vehicle for 48 h and then harvested. RNA was extracted from five paired samples and analysed with SurePrint G3 Human Gene Expression 8×60K v2 Microarray kit (Agilent Technologies, Tokyo, Japan). Altered gene expression was calculated as log2 (mRNA level of itraconazole- vs. vehicle-treated cells), and a difference of over two-fold (log2 ratio >1 or <−1) was considered as a change in the level of transcription.
Immunoblotting. Cell samples were lysed with Cellytic M lysis buffer containing Phosphatase Inhibitor Cocktail 2 and Protease Inhibitor Cocktail (all from Sigma-Aldrich). Proteins (2 μg) were size-fractionated by 15% or 5-20% sodium dodecyl sulfate–polyacrylamide gel electrophoresis and transferred to a polyvinylidene difluoride membrane (ATTO, Tokyo, Japan), which was blocked with EzBlock Chemi (ATTO) and incubated with primary antibodies against the following proteins: mTOR, phospho-mTOR, p70 S6 kinase (p70S6K), phospho-p70S6K, and AKT (all at 1:1,000 from Cell Signalling Technology, Danvers, MA, USA); and microtubule-associated protein 1A/1B-light chain 3 (LC3) and β-actin (both at 1:1,000; MBL, Nagoya, Japan). After washing with phosphate-buffered saline containing 0.01% Tween 20 (Wako Pure Chemical Industries, Osaka, Japan), the blots were probed with secondary antibodies for 1 h at room temperature. Immunoreactivity was visualized with an ECL Prime Western Blot Detection kit (GE Healthcare Life Sciences, Little Chalfont, UK). Band intensity was quantified on ImageQuant LAS4010 with ImageQuant TL software (GE Healthcare Life Sciences). The relative intensity of the target protein band was normalized to that of the internal control (β-actin) in each sample.
Statistical analysis. Differences between two groups were evaluated with the Mann–Whitney U-test using XLSTAT 2014 software (Addinsoft, Paris, France). Values of p<0.05 were considered significant.
Results
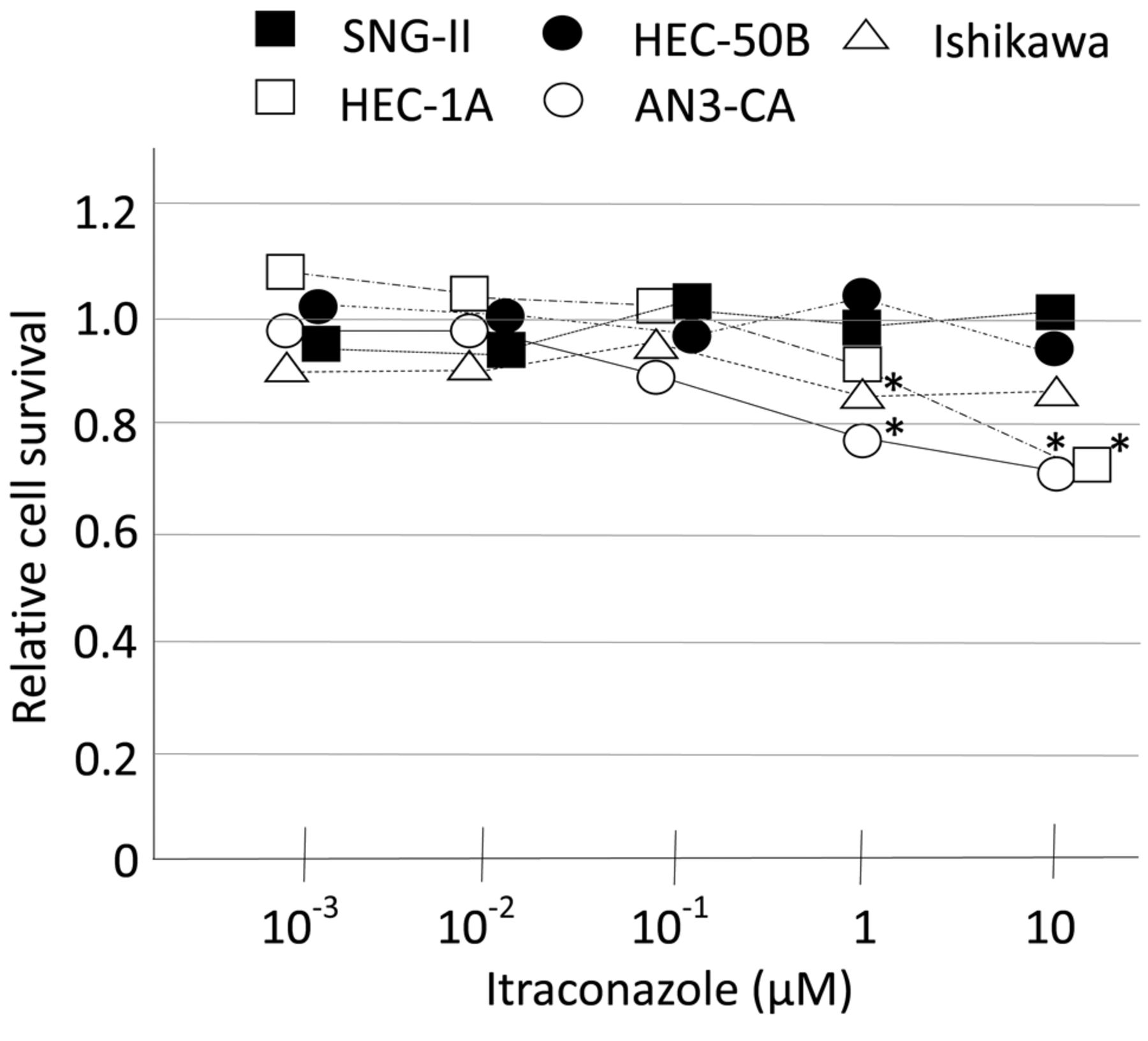
Itraconazole inhibits EC cell proliferation. Itraconazole treatment inhibited the proliferation of Ishikawa and AN3-CA cells at 1 μM and of HEC-1A and AN3-CA cells at 10 μM. SNG-II and HEC-50B cells were unaffected by itraconazole (Figure 1).
Itraconazole alters gene expression in EC cells. Treatment of EC cells with 10 μM itraconazole for 48 h increased GLI1 transcription by over 3-fold in Ishikawa and SNG-II cells (Table I). Key molecules involved in autophagy were unaffected by itraconazole treatment, except for Beclin 1, which was down-regulated in Ishikawa cells. The levels of the cholesterol-trafficking genes sterol carrier protein 2 (SCP2) and StAR-related lipid transfer domain protein 3 (STARD3) were unaltered by itraconazole, while the ATP-binding cassette transporter A1 (ABCA1) gene was down-regulated in Ishikawa, HEC-50B, and SNG-II cells. Transcription of ABCB1, encoding P-glycoprotein 1 (multidrug-resistance protein 1) and of ABCG1 encoding breast cancer resistance protein was unaffected by itraconazole treatment (Table I).
AKT/mTOR signaling is suppressed by itraconazole treatment. mTOR phosphorylation was reduced in the presence of 10 μM itraconazole relative to the control in all EC cell lines except Ishikawa cells (Figure 2). In AN3-CA and HEC-1A cells, in which proliferation was suppressed by itraconazole treatment, p70S6K and AKT phosphorylation were reduced after incubation for 48 h with 10 μM itraconazole (Figure 3). The expression of microtubule-associated protein 1A/1B-light chain (LC)3-II was increased after incubation for 48 h with 10 μM itraconazole in all cell lines examined, except for Ishikawa cells (Figure 4).
Discussion
Down-regulation of GLI1, a downstream effector of the Hedgehog signalling pathway, was not observed in the five EC cell lines examined in this study. Itraconazole treatment for 48 h increased GLI1 transcription by over 3-fold in Ishikawa and SNG-II cells. However, it suppressed mTOR signalling, with the exception of Ishikawa cells.
Genetic abnormalities and aberrant signalling vary between different histological subtypes of EC and even within the same subtype. An integrated genomic characterization of EC by The Cancer Genome Atlas Research Network revealed that mutations in phosphatase and tensin homolog or phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha and alterations in phosphatidylinositol 3-kinase (PI3K)/AKT signalling were common in EC (20). Moreover, EC cell lines were found to respond to PI3K/mTOR pathway inhibitors according to their mutations (21). However, phase II studies of mTOR inhibitors showed no correlation between response and the presence of PI3K/AKT pathway mutations (22). The mTOR inhibitor GDC-0941 was shown to suppress proliferation in Ishikawa, HEC-50B, and SNG-II cells, whereas temsirolimus had the same effect in HEC-1A and HEC-50B cells (21). In the current study, itraconazole suppressed proliferation of Ishikawa, AN3-CA, and HEC-1A cells, suggesting that its target is distinct from those of GDC-0941 and temsirolimus.
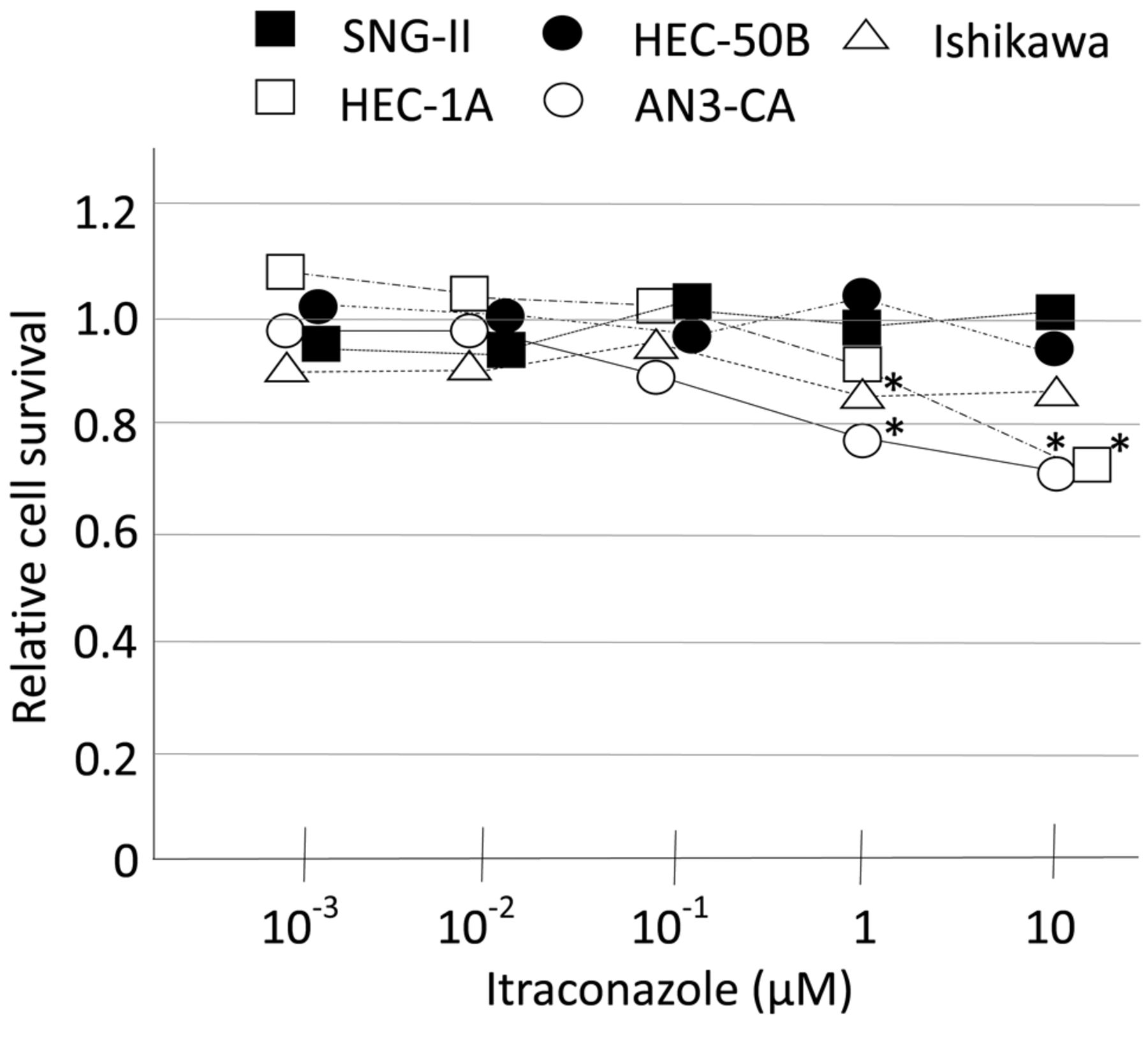
Proliferation of EC cells. Five EC cell lines were cultured with indicated concentrations of itraconazole for 48 h, and cell viability was evaluated with the MTT assay. Results were compared to those of the vehicle-treated control group with the Mann-Whitney U-test. *p<0.05.
Cholesterol is a major component of cell membrane microdomains known as lipid rafts, which contain a variety of signaling proteins and receptors (23). Changes in lipid composition have been reported in cancer cells, and therapeutic strategies that target lipogenic enzymes have been investigated in preclinical and clinical studies (24). Aberrant activation of AKT is correlated with an increase in lipid raft formation, while disruption of lipid rafts inhibits AKT activation. A previous study showed that itraconazole inhibited intracellular cholesterol trafficking to the plasma membrane in human umbilical vein endothelial cells, thereby suppressing mTOR signalling (9). Cholesterol transport was perturbed in human U87 glioblastoma cells treated with itraconazole; its redistribution was induced by down-regulation of SCP2 (11), which is located in multiple organelles, including mitochondria, and plays a key role in cholesterol transport (25, 26). In the current study, itraconazole had no effect on SCP2 and STARD3 transcription. ABCA1, which regulates cholesterol efflux across the plasma membrane, was down-regulated by 10 μM itraconazole treatment in Ishikawa, HEC-50B and SNG-II cells, in which proliferation was unaffected.
Itraconazole has been shown to induce autophagy and thereby inhibit the growth of glioblastoma cells (11). We observed here that protein expression of LC3-II, an autophagy marker, was up-regulated in all EC cell lines except Ishikawa in the presence of 10 μM itraconazole for 48 h. On the other hand, growth was suppressed in HEC-1A and AN3-CA cells under these conditions. These results suggest that autophagy is not correlated with proliferation in EC cells. However, there are currently no highly accurate assays for measuring autophagy-induced cell death in cancer cells (27, 28); this point requires further investigation.
Fold change in gene transcription induced by itraconazole treatment. Expression of key genes associated with the hedgehog signaling, autophagy, and lipid transport after incubation with 10 μM of itraconazole was calculated as log2 values relative to the mRNA level of vehicle-treated cells. Values representing >2-fold change (log2 ratio >1 or <−1) in expression are shown in bold.
Each of these cell lines harbours multiple mutations and shows cross-talk between different intracellular signalling pathways. It was reported that itraconazole inhibited Hedgehog and AKT/mTOR as well as WNT/β-catenin signalling (29). Our results suggest that in EC cells, itraconazole interferes with AKT/mTOR signals. We are currently carrying out a window-of-opportunity clinical trial to assess the anticancer activity of itraconazole and identify biomarkers in responders (UMIN000018388).

Itraconazole suppresses mTOR signaling. Cells were cultured with 10 μM itraconazole (Itra) for 24, 48, and 72 h and with vehicle for 24 h; protein expression was then analyzed by western blotting.

Itraconazole suppresses AKT and p70S6K (S6K) phosphorylation. Cells were incubated with itraconazole (Itra) for 48 h and phosphorylation of AKT (p-AKT) and p70S6 kinase (p-S6K) was evaluated by western blotting.

Itraconazole (Itra) treatment induces LC3-II expression.
Acknowledgements
Ishikawa human EC cells were a gift from Dr. Masato Nishida (Tsukuba University, Ibaraki, Japan). This work was supported by a Japan Society for the Promotion of Science KAKENHI grant (no. JP16K11166 to H. Tsubamoto) and Grant-in-Aid for Researchers, Hyogo College of Medicine, 2016 (to K. Inoue).
- Received November 12, 2016.
- Revision received December 20, 2016.
- Accepted December 22, 2016.
- Copyright© 2017, International Institute of Anticancer Research (Dr. George J. Delinasios), All rights reserved
References
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Itraconazole Inhibits Intracellular Cholesterol Trafficking and Decreases Phosphatidylserine Level in Cervical Cancer Cells
- Chloroquine Enhances Rapamycin-induced Apoptosis in MG63 Cells
- Itraconazole Modulates Hedgehog, WNT/{beta}-catenin, as well as Akt Signalling, and Inhibits Proliferation of Cervical Cancer Cells