


Abstract
Background/Aim: Recent reports have indicated that hyperglycaemia is associated with breast cancer progression. High glucose conditions corresponding to hyperglycaemia significantly promote migration of MCF-7 human breast cancer cells, however, little is known about the mechanisms of glucose sensing for the acquisition of migratory properties by MCF-7 cells. This study investigated glucose sensing and mediation, which are responsible for the high motility of MCF-7 cells. Materials and Methods: We evaluated the migration of MCF-7 cells cultured in high glucose-containing medium and essential regulatory factors from the perspective of the glucose transport system. Results: We demonstrated that glucose transporter 12 (GLUT12) protein level increased in MCF-7 cells and co-localized with actin organization under high glucose conditions. Moreover, GLUT12-knockdown completely abrogated high glucose-induced migration, indicating that GLUT12 functionally participates in sensing high glucose concentrations. Conclusion: GLUT12 plays a critical role in the model of breast cancer progression through high glucose concentrations.
Women with diabetes have a worse survival than those without diabetes, and diabetes was recently demonstrated to be closely related to breast cancer prognosis (1-9). Moreover, clinical epidemiological studies have shown that breast tumor progression and response to therapy differ between women with and without diabetes (4, 7, 10). The presence of a high glucose level, corresponding to hyperglycemia in vivo, has been reported to modulate breast cancer cell functions including survival, growth, and proliferation, suggesting the glucose level as being critical for breast cancer progression in patients with diabetes (11-19). Our previous studies also demonstrated that the migratory ability of MCF-7 human breast cancer cells, which is linked to invasion and metastasis, was strongly dependent on glucose concentration (13, 14). However, the mechanisms responsible for detecting changes of glucose level leading to changes in breast cancer development are largely unknown. Therefore, studies aiming to understand the essential regulatory factors in breast cancer progression in individuals with diabetes are necessary to accelerate the development of potential targets for cancer therapies.
Extracellular glucose is sensed and transported into cells by two classes of hexose transporters: the sodium-dependent glucose transporter (SGLT) family and the facilitative-type glucose transporter (GLUT) family (20-23). These glucose transporters exhibit cell-specific expression and specialized glucose-sensing and responding properties which contribute to glycolysis and related cellular functions (20-23). It has been demonstrated in many tumor cells, including breast cancer cells, that utilization of glucose is tumor stage-dependent and is associated with an altered expression of glucose transporters (22-26). SGLT1 expression has been reported to be positively related to the expression of epidermal growth factor receptor (EGFR) in cancer cells, suggesting that EGFR functions linked to glucose transport system may be related to resistance of tumor cells to chemotherapeutic agents and tyrosine kinase inhibitors (27). Moreover, altered expression and localization of GLUTs 1-6 has been detected on stimulation with estradiol, EGF and hypoxic stress, in malignant breast cancer cells using RNA analysis or immunohistochemistry, indicating they participate in invasive potential and affect the survival of patients with breast cancer (20, 24, 26, 28, 29). Most interestingly, GLUT12 was identified and cloned from the human breast cancer cell line, MCF-7, and this transporter was found to be localized within the cells, in particular at the perinuclear regions (24, 30). Trafficking of GLUT12 to the cell surface and regulation of GLUT12 expression aggressively participate in breast cancer development, therefore, targeting of GLUT12 could be a novel strategy in diagnosis and treatment of breast cancer (24, 31). However, the functional importance of glucose transport systems in high glucose level-induced breast cancer has not yet been elucidated in detail, and its evaluation is required in order to develop sophisticated strategies for effective breast cancer therapy in patients with diabetes.
In this study, we investigated the glucose-sensing mechanisms in MCF-7 cells cultured under hyperglycemia.
Materials and Methods
Cell lines and cell cultures. MCF-7 and MDA-MB-231 Human breast cancer cells, obtained from the European Collection of Cell Cultures (Salisbury, UK), were grown as monolayer in minimum essential medium (MEM; Invitrogen, Carlsbad, CA, USA) containing 5.5 mM D-glucose supplemented with 10% heat inactivated fetal bovine serum (FBS; Corning, Riverfront, NY, USA), 100 U/ml penicillin and 100 μg/ml streptomycin (Nacalai Tesque, Kyoto, Japan) at 37°C in a humidified and equilibrated (5% CO2, 95% air) incubator.
To investigate the migratory behavior under high glucose levels corresponding to hyperglycemia, MCF-7 and MDA-MB-231 cells were cultured in media with a normal (5.5 mM) or high (25 mM) D-glucose concentration, or in osmotic control media (5.5 mM D-glucose plus 19.5 mM D-mannitol) with 10% FBS.
Scratch wound-healing assay. Scratch wound-healing assays were performed as described previously (13). A confluent monolayer of cells cultured with MEM containing 10% FBS and 5.5 mM D-glucose in a 6-well plate was scratched with a sterile 10 μl pipette tip to create a straight line in each well. After scratching, the wells were gently washed with phosphate buffered-saline (PBS) to remove the debris, and then the medium was replaced with MEM containing 10% FBS, 5.5 mM or 25 mM D-glucose, or osmotic control (5.5 mM D-glucose plus 19.5 mM D-mannitol) in the presence or absence of 25 mM for the glucose analog 2-deoxy-D-glucose (2-DG; Nacalai Tesque), 1 μM for GLUT inhibitor cytochalain B (Nacalai Tesque), 100 μM for GLUT inhibitor phloretin (Sigma-Aldrich, St. Louis, MO, USA) and 100 μM for SGLT inhibitor phloridzin dihydrate (Sigma-Aldrich). The cells were incubated for 12-24 h at 37°C in a humidified and equilibrated (5% v/v CO2) incubator. The images of wounded areas at 0 and 12-24 h after scratching were acquired using a microscopy system (Nikon, Tokyo, Japan) and the cell-free wound area was quantified by using ImageJ software (ver. 1.46; NIH Image, Bethesda, MD, USA). The migratory rate was evaluated by calculating the proportion of the closed area from the initial time to 12-24 h-points. Multiple views of each well were examined, and at least three independent experiments were performed.
Transwell migration assay. Transwell migration assays were carried out using 8.0 μm pore size Costar Transwell inserts (Corning Inc., Tewksbury, MA, USA) as described previously (13). Cells were seeded on the upper chambers of 24-well transwell plates at 2.5×104 cells and were cultured in MEM containing 10% FBS with D-glucose (5.5 mM or 25mM), or osmotic control (5.5 mM D-glucose plus 19.5 mM D-mannitol). After 24 h incubation, the motile cells at the bottom of the filter were fixed with 4% formaldehyde and stained with 4’,6’-diamidino-2-phenylindole (DAPI; Sigma-Aldrich). The number of migrating cells was quantified by counting the stained cells under a Nikon fluorescent microscopy system.
Western blot analysis. The treated and untreated cells were lysed with lysis buffer containing 100 mM NaCl, 2.5 mM HEPES, 2 w/v% CHAPS (Nacalai Tesque) and a protease inhibitor cocktail (Roche Diagnostics GmbH, Mannheim, Germany) and centrifuged at 17,360 × g for 10 min at 4°C. The protein concentration of the cell lysates was determined using a QuantiPro™ BCA Kit (Sigma-Aldrich). From each sample, 20 μg protein in the supernatant was separated by 10% polyacrylamide gel and then blotted onto a Immobilon® Transfer Membranes (Merck Millipore, Darmstadt, Germany). After blocking with 5% bovine serum albumin (Sigma-Aldrich) in PBS containing 0.1% Tween 20 (PBS-T; Wako, Tokyo, Japan) for 90 min at room temperature, the membranes were incubated with primary antibodies overnight at 4°C. The following primary antibodies and corresponding dilutions were used in the experiment: rabbit polyclonal antibody to GLUT12 (1:500; Abcam, Cambridge, UK); rabbit polyclonal antibody to GLUT1 (1:1000; Abcam), and mouse monoclonal antibody to glyceraldehyde 3-phosphate dehydrogenase (GAPDH; 1:1,000; Santa Cruz Biotechnology, Inc., Dallas, TX, USA). After washing with PBS-T, the membranes were incubated with horseradish peroxidase-conjugated secondary antibodies (GE Healthcare, Buckinghamshire, UK) to rabbit IgG (1:2,000 dilution) and mouse IgG (1:1,000 dilution), as appropriate, for 1.5 h at room temperature. The bands were detected by enhanced chemiluminescence using ECL Western Blotting Detection Reagents (GE Healthcare). The western blotting results were quantified by densitometric analysis with ImageJ software (ver. 1.46; NIH Image), and the relative expression levels of the above proteins were normalized to those of GAPDH, which was used as an internal control.
Immunohistochemistry. MCF-7 cells were grown on 13 mm coverslip at a density of 1.5×105 cells/well. The following day, the medium was replaced with MEM containing 10% FBS with normal (5.5 mM) or high (25 mM) D-glucose concentration, or osmotic control (5.5 mM D-glucose plus 19.5 mM D-mannitol). After 1-h incubation, the cells were washed with PBS, fixed with 4% paraformaldehyde for 30 min and permeabilized with 0.2% Triton X-100 for 10 min. After blocking with 5% bovin serum albumin in PBS for 30 min, the cells were incubated with primary antibody to GLUT12 (Abcam) overnight at 4°C. After further washing, the cells were incubated with the Alexa Fluor® 488 labeled-secondary antibodies (Invitrogen) for 1 h at room temperature. To detect cellular F-actin, the cells were stained with Alexa Fluor® 546 labeled phalloidin (Invitrogen) for 90 min at room temperature and were then washed with PBS and analyzed using a Nikon fluorescent microscopy system (Tokyo, Japan).
Generation of GLUT12-knockdown in MCF-7 cells. Human GLUT12 MISSION® siRNA (NM_145176; Sigma-Aldrich) and scrambled MISSION® Universal Negative Control siRNA (SIC001; Sigma-Aldrich) were transfected into MCF-7 cells using Lipofectamine 2000 Reagent (Invitrogen) according to the manufacturer's protocol. After 48 h of transfection, the cells were collected for western blot analysis or used for transwell migration assays.
Statistical analysis. All statistical analyses were performed using GraphPad Prism (ver. 5.00; GraphPad, San Diego, CA, USA). For multiple-group comparisons, either one-way analysis of variance (ANOVA) followed by Tukey's post hoc test or two-way ANOVA followed by Bonferroni's post hoc test was performed. Differences were considered significant when the calculated p-value was less than 0.05.
Results
High glucose levels promote motility of MCF-7 human breast cancer cells through the glucose transport system. We investigated the effects of high glucose levels on the motility of estrogen receptor-positive MCF-7 and estrogen receptor-negative MDA-MB-231 human breast cancer cell lines using a scratch wound-healing assay and transwell migration assay (Table I). The rate of migration into the scratch wound area was significantly promoted in MCF-7 cells exposed to medium containing 25 mM glucose (hyperglycemic level) by about 1.7-fold compared with those exposed to 5.5 mM glucose (normal physiological glucose level) and 5.5 mM glucose and 19.5 mM mannitol (osmotic control). In the transwell migration assay, MCF-7 cells cultured with 25 mM glucose medium showed high migratory activity (Table I), demonstrating similar results with those of the wound-healing assay. On the other hand, MDA-MB-231 cells showed no increase in migration under high glucose conditions. These findings are consistent with a previous report on the association between extracellular glucose concentration and migratory ability (13), and suggest that a specific and glucose-related molecule expressed in MCF-7 cells but not MDA-MB-231 cells might play a critical role in high glucose-induced migration.
Next, we examined the effects of specific inhibitors of glucose metabolism and glucose transporters on migratory ability to clarify the glucose-sensing and transducing mechanisms responsible for the high motility of MCF-7 cells cultured in high glucose medium. Treatment with 2-DG, a non-metabolizable glucose analog inhibiting phosphorylation of glucose by hexokinase in the first step of glycolysis, was not able to inhibit high glucose-induced migration, indicating that glucose metabolism is independent of glucose sensing and migration (Figure 1A). Moreover, regarding glucose transporters, phloridzin, an inhibitor of SGLT isoforms (23), had no effect on high glucose-induced migration (Figure 1B). On the other hand, the promotion of migration under high glucose conditions was significantly inhibited by cytochalasin B, an inhibitor of glucose binding to the cytoplasmic domains of all GLUT isoforms (23) (Figure 1C), and phloretin, a inhibitor of glucose binding to the extracellular domain of all GLUT isoforms (23) (Figure 1D). These findings demonstrate that the promotion of migration under high glucose levels is facilitated not by SGLT but the GLUT glucose transporter system in MCF-7 cells.
Effects of a high glucose level on migration of breast cancer cells. MCF-7 and MDA-MB-231 cells were cultured with normal D-glucose (Glc) medium (5.5 mM), high Glc medium (25 mM), or osmotic control medium [5.5 mM Glc plus 19.5 mM D-mannitol (Man-ol)] for 24 h, and the number of migrated cells were analyzed as described in the Materials and Methods. Data are presented as means±SD.
GLUT12 as glucose sensor and mediator in high glucose-induced migration. To investigate the mechanism of high glucose-promoted migration through GLUT in MCF-7 cells, we focused on GLUT12 as a glucose sensor and mediator. It has been reported that GLUT12 is specifically expressed in estrogen receptor-positive cells MCF-7 and tightly regulates glucose uptake and cell functions according to stimulation by hormones and stress (such as estradiol, EGF and hypoxia) (24). We found that GLUT12 was expressed in estrogen receptor-positive cells (MCF-7), but was not detected in estrogen receptor-negative cells (MDA-MB-231) (Figure 2A). Moreover, the expression of GLUT12 protein was markedly increased after 1 h treatment with high glucose compared with 5.5 mM glucose or 5.5 mM glucose and 19.5 mM mannitol, and decreased after 24 h (Figure 2B). On the other hand, there were no significant differences in the GLUT1 expression level among treated groups and times in MCF-7 cells (Figure 2B). To further characterize the expression of GLUT12 in response to glucose stimulation in MCF-7 cells, immunohistochemistry was performed. GLUT12 was intracellularly located and at the perinuclear region in cells cultured with 5.5 mM glucose (Figure 3A). A high glucose level significantly increased the proportion of cells with punctate intracellular and cell surface formations of GLUT12 compared with those treated with 5.5 mM glucose, or 5.5 mM glucose and 19.5 mM mannitol (Figure 3A and B). We also demonstrated the co-localization of GLUT12 with crowding and organization of F-actin in cells cultured in 25 mM glucose (Figure 3C), indicating that GLUT12 is associated with high glucose-induced migration in MCF-7 cells.
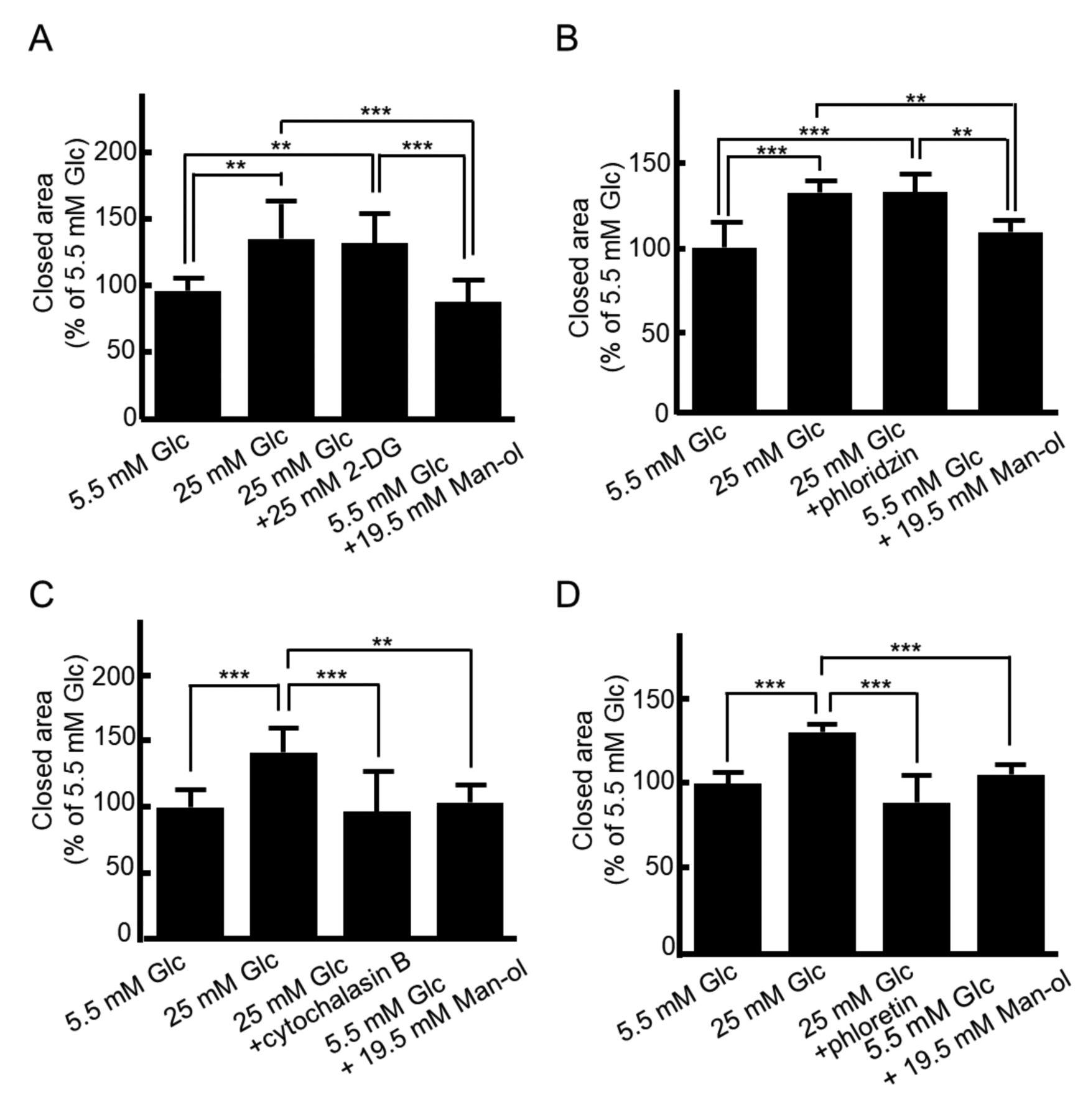
High glucose-induced migration is mediated through the GLUT glucose transporter system in MCF-7 cells. A confluent monolayer of MCF-7 cells was scratched and then cultured in normal D-glucose (Glc) (5.5 mM) medium, high Glc (25 mM) medium, or osmotic control medium [5.5 mM Glc plus 19.5 mM D-mannitol (Man-ol)] in the presence or absence of 25 mM Glc analog 2-deoxy-D-glucose (2-DG) for 12 h (A), 100 μM sodium glucose transporter (SGLT) inhibitor phlorizin (B), 1 μM GLUT inhibitor cytochalasin B (C) or 100 μM GLUT inhibitor phloretin (D) for 24 h. The wounded area was then photographed under microscopy. The data are presented as the mean±SD. Significantly different at ***p<0.001, n=5-9.

A high glucose level regulates glucose transporter 12 (GLUT12) protein level in MCF-7 cells. MCF-7 cells were cultured with normal D-glucose (Glc) (5.5 mM) medium, high GIc (25 mM) medium, or osmotic control medium [5.5 mM GIc plus 19.5 mM D-mannitol (Man-ol)) for 1 or 24 h. The changes in GLUT1 and GLUT12 protein levels were assessed by western blot (A, B) and quantitative analysis (B lower panel). The data are presented as the mean±SD. Significantly different at *p<0.05, **p<0.01. n=3.
To further understand the function of GLUT12 in high glucose-induced migration, we examined GLUT12 knockdown in MCF-7 cells using siRNA specific for human GLUT12. GLUT12 knockdown significantly reduced GLUT12 protein levels in MCF-7 cells compared to control cells, as demonstrated by western blot analysis (Figure 4A). As shown Figure 4B, the high migratory activity of MCF-7 cells in medium with 25 mM glucose was completely abrogated by GLUT12 knockdown. These results suggest that GLUT12 plays an essential role in the promotion of MCF-7 motility following stimulation with a high glucose level.

Intracellular localization of glucose transporter 12 (GLUT12) in MCF-7 cells cultured with high glucose level. MCF-7 cells were cultured with D-glucose (Glc) (5.5 mM) medium, high Glc (25 mM) medium, or osmotic control medium [5.5 mM Glc plus 19.5 mM D-mannitol (Man-ol)] for 1 h. Images (A) and quantitative analysis (B) of positive cells with punctate formations of GLUT12 are shown under each experimental condition. Scale bars represent 10 μm. The data are presented as the mean±SD. **Significantly different at p<0.01, n=3. C: Photomicrographs showing GLUT12 staining (green), F-actin staining (red), and the merged image (green and red). Cells were exposed to high Glc concentration for 1 h. Scale bars represent 50 μm. Arrows show representative co-localization of GLUT12 and F-actin. Representative results of three independent experiments are shown.

Glucose transporter 12 (GLUT12) is essential for high glucose-stimulated migration of MCF-7 cells. A: MCF-7 cells were transfected with siRNA for GLUT12 or control sequences. The knockdown effect on GLUT12 was shown in western blotting. B: In Transwell chamber assays, MCF-7 cells transfected with siRNA for GLUT12 were incubated in medium with normal D-glucose (Glc) (5.5 mM) medium, high Glc (25 mM) medium, or osmotic control medium [5.5 mM Glc plus 19.5 mM D-mannitol (Man-ol)] for 24 h, and migrated cells were analyzed as described in the Materials and Methods. The data are presented as the mean±SD. Significantly different at *p<0.05, **p<0.01. n=3.
Discussion
In this study, we demonstrated that GLUT12 expression is required for the promotion of high glucose-stimulated cell motility. Although the migratory potential of cancer cells has been reported to be correlated with GLUT1 expression, we found that high GLUT1 expression was shown in both MCF-7 and MDA-MB-231 cells, and there were no significant differences in the GLUT1 expression level among treatments and times in MCF-7 cells, thus high glucose-induced migration in MCF-7 cells is not fully explained by GLUT1 alone, implicating the existence of novel glucose transporter system or glycolysis in breast cancer progression under high glucose conditions. Our findings indicate that high glucose-induced migration is not dependent on the glucose metabolic system and tightly regulated by GLUT12 of the glucose transporter system. GLUT12 expression was observed in MCF-7 cells but was not detected in MDA-MB-231 cells, in agreement with a previous report, demonstrating expression in estrogen receptor-positive cells (24). Our data also indicate that GLUT12 protein levels were significantly increased in MCF-7 cells treated with high glucose for 1 h, and GLUT12 knockdown blocked high glucose-induced migration. A previous report demonstrated that a transient increase in GLUT12 protein by estradiol is related to increased half-life of GLUT12 protein and plays an important role in early-stage breast cancer progression (24). GLUT12 expression levels may also be sophisticatedly regulated in high glucose conditions and mediated the first step of high glucose-induced migration in our model. Moreover, the intracellular localization of GLUT12 was found to be altered under a high glucose level, and we observed co-localization of GLUT12 with F-actin. The localized polymerization of actin cytoskeleton is known to form protrusive structures as a driving force for the invasive and metastatic phenotypes of malignant cancer cells, namely filopodia, lamellipodia, and invadopodia/podosomes (32, 33). We also demonstrated that a high glucose level induces alterations in cell shape and actin polymerization, and significant lamellipodia formation was also observed for cells treated with a high glucose level (13). Thus, it is possible that GLUT12 senses the extracellular glucose concentration and co-regulates proteins involved in the actin cytoskeleton, resulting in increased cell motility at high glucose levels, although further studies are required in an invasive model.
We previously showed that zinc transported via zinc transporters ZIP6 and ZIP10 play an important role in glucose-induced cell migration (13, 34). Zinc is a novel intracellular second messenger, and it is thought that this zinc system and GLUT12 synchronize and regulate the cell motility under stimulation with a high glucose level. Moreover, GLUT12 is proposed to play central roles regarding the cell motility signal pathway complex, the transportsome which allows the cooperation of multiple transporters under high glucose levels.
Recent reports have suggested that GLUT12 could potentially be a key protein in breast cancer progression (24). A large study of human breast cancer demonstrated expression of GLUT1 in 42% of tumors, suggesting the involvement of other glucose transporters (35), whilst GLUT12 immunostaining was shown in invasive tumors and non-invasive ductal carcinoma but no staining was shown in adjacent normal breast tissue (31). GLUT12 expressed in the non-invasive ductal carcinoma in situ component has been associated with early stage acquisition of an invasive phenotype in breast cancer (31). The present study demonstrates that GLUT12 functionally participates in sensing high glucose levels and is essential for glucose-induced cell migration. We suggest that GLUT12 may be a candidate marker for metastatic spread in hyperglycemia in patients with diabetes, although further studies of molecular mechanisms will be required. The elucidation of the role of GLUT12 in high glucose-induced cell migration could lead to new strategies for the diagnosis and therapy of breast cancer in patients with hyperglycemia.
Acknowledgements
This work was supported by a Nagai Memorial Research Scholarship from the Pharmaceutical Society of Japan (to C.M.) and a Grant-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (Grant Number 15K07955 to T. T-N.). The Authors thank Ms. Satomi Kawahara (Department of Pharmaceutics, School of Pharmacy and Pharmaceutical Sciences, Mukogawa Women's University, Hyogo, Japan) for her experimental support.
Footnotes
↵This Article is freely accessible online.
Disclosures
The Authors have no conflicts of interest in regard to this study.
- Received September 7, 2017.
- Revision received October 5, 2017.
- Accepted October 9, 2017.
- Copyright© 2017, International Institute of Anticancer Research (Dr. George J. Delinasios), All rights reserved
References
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.