


Abstract
Background/Aim: Heat shock protein 90 (HSP90) inhibitors have recently been tested as anticancer drugs in a variety of carcinomas. Yet, there exist only few reports about HSP90 inhibitor and its thepeutic effect on liposarcoma. The therapeutic effects of HSP90 inhibitors have been mainly observed in oncogenic and tumor angiogenic signaling cascades by observing tumor growth. Materials and Methods: We used the the LPS 246 liposarcoma cell line and GS-076 PDC (patient-derived cell lines). On these, we performed cell viability assays and migration assays under treatment with the HSP90 inhibitor, 17AAG. For analyzing angiogenesis factor, we used quantitative polymerase chain reaction (qPCR) after treating cells with the 17AAG inhibitor. Regarding in vivo assay, we made the tumor model in immune-deficient mouse and compared the tumor size of drug-treated group at each time point with controls. For sequestering analysis of angiogenesis factor in vivo, we performed immuno-fluorescence (IF) staining on tumor tissue. Results: Through cell viability, migration assay and qPCR about angiogenesis factor, we demonstrated the anti-oncogenic and anti-angiogenic effects of an HSP90 inhibitor on a liposarcoma cell line and a patient-derived primary cell model (PDC). Also, the HSP90 inhibitor 17AAG effectively inhibited the activity of protein kinase B (AKT) and blocked extracellular signal-regulated kinase (ERK) activity. Hence, 17AAG effectively disrupted the oncogenic signaling cascade and substantially inhibited tumor growth in vitro. In an LPS 863 cell xenograft mouse model treated with 17AAG, we observed that tumor size was decreasing, as well as down-regulation of the expression levels of vascular endothelial growth factor receptor 2 (VEGFR2), CD31 and signal transducer and activator of transcription-3 (STAT3). Conclusion: 17AAG reduced the activity of AKT, ERK, VEGF and STAT3 in oncogenic and angiogenic pathways in liposarcoma PDC models derived from patients' tissues and cancer cell lines.
Liposarcoma is a common type of soft tissue sarcoma. It accounts for 10% of pediatric sarcoma patientsand 8% of adolescent and adult sarcoma patients(1). Although the primary treatment mainly consists of a surgical resection, if a relapse or a metastatic event has occurred owing to an incomplete surgical resection, malignant tumors may occur (2). While chemotherapy is a useful therapy for malignant liposarcoma patients, patients with metastases have a poor prognosis with less than a 25% disease-free survival rate (3). Therefore, the development of new treatment strategies is necessary.
Current first-line chemotherapy treatments include the single agent use of anthracyclines (doxorubicin) or an anthracycline-based combination product (doxorubicin and ifosfamide or dacarbazine). Second line treatment involves non-anthracycline combinations (gemcitabine and docetaxel) (4). Recently, flavopiridol has been suggested as a potent alternative adjuvant agent for liposarcoma treatment (4-6). Heat shock protein 90 (HSP90) has been observed to be 2-10-fold over expressed in tumor tissue compared to normal tissue (1, 7, 8). It has also been described that liposarcoma leads to overexpression in human tumor tissues (9). Importantly, signal transducers (STAT3), intracellular kinases (ARK, ERK) and growth factor receptors (MET) are common oncogenic transcription factors and signaling intermediates of HSP90 client proteins (9, 10).
HSP90 is a protein that maintains the functionality and integrity of a wide range of various oncogenic proteins (11). HSP90 inhibitors have recently been tested regarding their effectiveness as anticancer drugs in a variety of carcinomas, including liver cancer (12), stomach cancer (13), head and neck squamous cell carcinoma (HNSCC) (14), and breast cancer (15). However, little research has been carried out to support the argument that targeting by an HSP90 inhibitor would have therapeutic effects on liposarcomas (16). Inhibition of HSP90 caused anti-angiogenic effects in the present study. The major targets were confirmed as VEGFR2 and platelet-derived growth factor receptor-β (PDGFR-β) in endothelial cells (ECs) and vascular smooth muscle cells (VSMCs), respectively (18). Here, we suggest that AKT and ERK are the major targets in the tumorigenesis of liposarcoma, and an HSP90 inhibitor may be an effective anti-angiogenic agent by blocking the interleukin-6 (IL-6)-STAT3-VEGF pathway in engrafted tumor tissues of mice models.
Materials and Methods
Cell culture. A de-differentiated human liposarcoma cell line, LPS863, was provided by Dr. Dina Lev (MD Anderson Cancer Center, Holcombe Blvd, Houston, TX. USA). 14GS-076, which is derived from liposarcoma patient tissues, was established by the Sarcoma Research Center at the Samsung Medical Center in Korea. The cells were maintained in Dulbecco's modified Eagle's medium (DMEM) HyClone, South Logan, UT, USA) supplemented with 10% fetal bovine serum. The HSP90 inhibitor 17AAG was purchased from Santa Cruz Biotechnology (Dallas, TX, USA) and dissolved in PBS.
Cell viability. WST1 (Clonetech, Kyoto, Japan) and a CellTighter-Fluor™ kit (Promega, Madison, WI, USA) were used to measure the relative number of live cells in a culture population after experimental manipulation. All procedures were performed according to the manufacturer's instructions. Briefly, cells seeded in wells of an opaque 96-well microtiter plate (5×103 cells/well) were incubated with PBS (as a control group) or 500 nM 17AAG for indicated time-points. The plate was incubated under standard cell culture conditions. Tests were performed in triplicate; three wells with medium only served as blanks and two wells with untreated cells served as controls. The plate was loaded into a dispensing fluorescence detector (Multiplate Reader, Mithras2, Berthold, Bad, Wildbad, Germany) and the resulting fluorescence levels were analyzed by adding 10 μL of the WST1 mixture or 100 μL of the Cell Tighter-Fluor™ mixture and releasing reagent to each well followed by incubation for at least 30 minutes at 37°C. The relative light units at 340-380 nm/505 nm (WST1: 450 nm) were measured. Statistical analysis was performed using an unpaired Student's t-test.
Migration assay. The CytoSelect™ Wound Healing kit (Cell Biolabs, INC., San Diego, CA, USA) was used to examine the migration levels of liposarcoma cell treated with 17AAG. According to the provided procedure, we measured migration levels of each treated cell line in triplicate. Briefly, using sterile forceps, the desired numbers of inserts were oriented in the plate wells with their “wound field” aligned in the same direction. A cell suspension containing 1.0×106 cells/ml in media was created with 10% fetal bovine serum (FBS). Cells were incubated in a cell culture incubator overnight or until a monolayer formed, and the insert was carefully removed from the well to begin the wound healing assay. Next, we measured the percent closure or the migration rate of the cells into the wound field. Migrated Area (Percent of Closure, %)=Migrated Cell Surface Area/Total Surface Area ×100.
Western blotting. Whole-cell lysates from LPS863 and 14GS-076 cells, as well as from subcutaneous tumors, were prepared as previously described (19). Protein samples (20 μg) were subjected to western blotting via denaturing 10% SDS-PAGE electrophoresis. Membranes were probed with antibodies against phosphor-AKT (Ser473), AKT, phosphor-ERK (Thr202), total-ERK, phosphor-STAT3(Tyr705), total-STAT3, CD31 and β-ACTIN (Sigma, Lewis, MO, USA). Antibodies were detected by enhanced chemilu-minescence (Young-In, Seoul, Republic of Korea). All of procedures were performed in triplicate.
Detection of IL-8 mRNA expression by real-time quantitative PCR. Total RNA preparation and the RT reaction were carried out as described previously (20). PCRs were performed using an ABI Prism 7900HT according to the manufacturer's instructions. Amplification of specific PCR products was detected using the SYBR Green PCR Master Mix (Applied Biosystems). The primers designed in this study were as follows: IL-8 forward primer, 5’-ACT GAG AGT GAT TGA GAG TGG AC-3’; IL-8 reverse primer, 5’-AAC CCTCTG CAC CCAGTT TTC-3’; VEGF forward primer, 5’-TCACCGCCTCGGCTTGTCACAT-3’; VEGF reverse primer, 5’-GAGATGAGCTTCCTACAGCAC-3’; IL-6 forward primer, 5’-CACAGACAGCCACTCACCTC-3’; IL-6 reverse primer, 5’-TTTTCTGCCAGTGCCTCTTT-3’; GAPDH forward primer, 5’-CGG CGACGA CCC ATT CGA AC-3’; and GAPDH reverse primer, 5’-GAA TCGAACCCT GAT TCC CCG TC-3’. RNA samples were normalized to the level of GAPDH mRNA. The real-time PCR was performed in triplicate in a total reaction volume of 25 μL containing 12.5 μL of SYBR Green PCR Master Mix, 300 nM forward and reverse primers, 11 μL of distilled H2O, and 1 μL of cDNA from each sample. Samples were heated for 10 min at 95°C and amplified for 40 cycles of 15 sec at 95°C and 60 sec at 60°C. Quantification was performed using the 2−ΔΔCt method, where the Ct value was defined as the threshold cycle of PCR at which the amplified product was detected. The ΔCt was obtained by subtracting the housekeeping gene (GAPDH) Ct value from the Ct value of the gene of interest (IL-8, VEGF, and IL-6). The present study used the ΔCt of control subjects as the calibrator. The fold change was calculated according to the formula 2−ΔΔCt, where ΔΔCt was the difference between ΔCt and the ΔCt calibrator value (which was assigned a value of 1 arbitrary unit).
Xenograft tumor models. LPS863 cells (2×107 cells) were subcutaneously injected into immune-deficient mice, NOD/Shi-scid IL-2Rγnull (JAXON Lab, Bar Harbor, ME, USA). Mice were randomized and assigned to treatment groups and intraperitoneally injected with 100 μl, 3 times a week, with diluents of 17AAG (25 mg/kg), starting on the day when the tumor was palpable on the mouse. The experiment was terminated after 5 weeks. All of the procedures were approved and performed according to the guidance of Samsung animal and use committee in Korea.
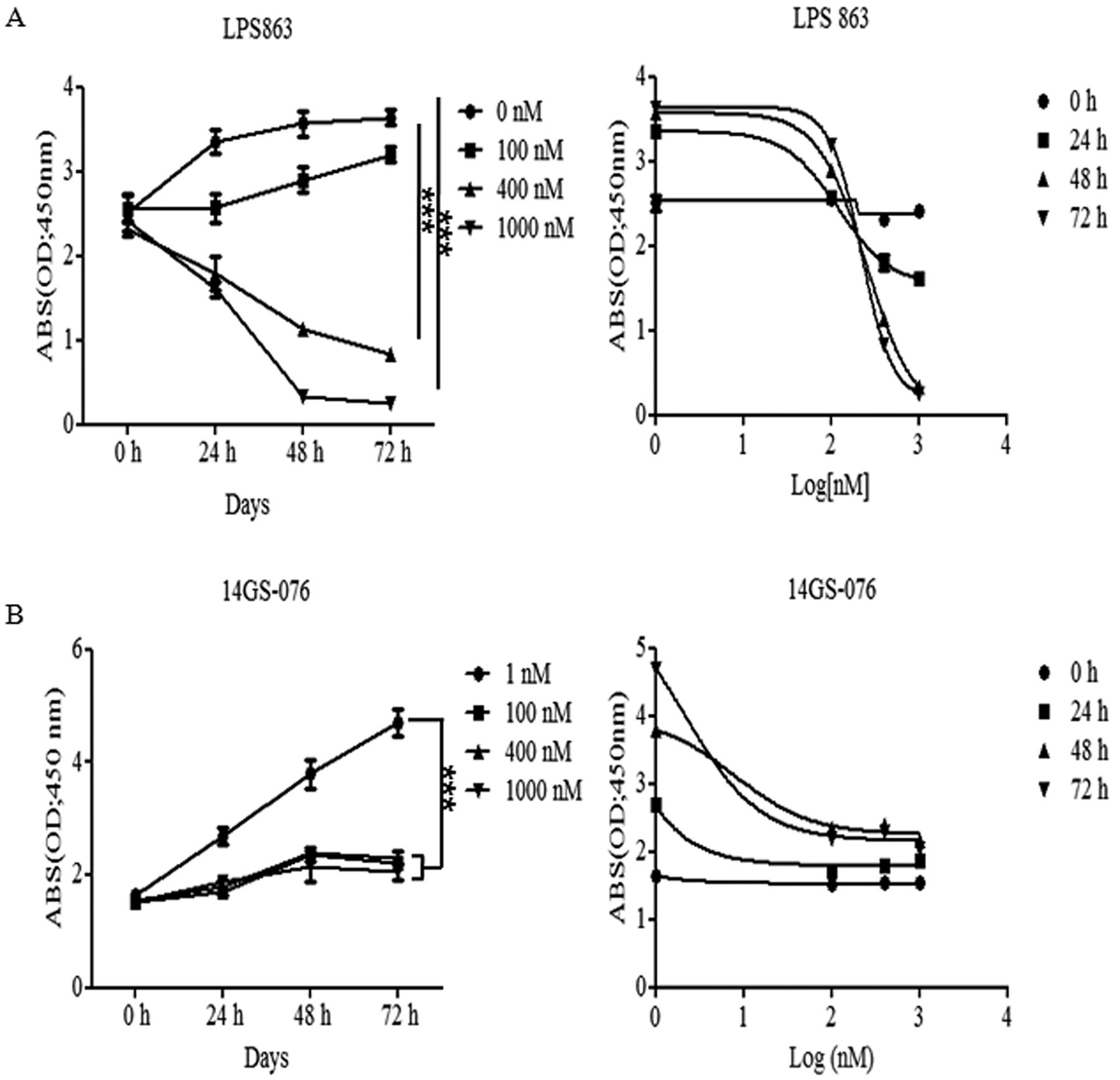
Effect of HSP90 inhibitor 17AAG on liposarcoma cells.LPS863 (A) and 14GS-076 cells (B) were treated with different concentrations of 17AAG (0-1,000 nM) and different time points (0, 24, 48 and 72 h). Proliferation and viability were determined using the WST1 assay. IC50 was determined at 500 nM, dose of 17AAG on both of cells. Data are shown relative to PBS-treated controls. The results are expressed as percentages of control cells. Each bar is the mean±SD from one experiment performed in triplicate. ***p<0.0005.
PET analysis. These animals were divided into 2 groups of 5 mice each. Both groups were injected with 18F-FDG in the tail vein. At 1h injection, the animals were sedated using isoflurane and imaged with a microPET camera (Concorde Microsystems Inc., Cogdill Road, Knoxville, TN, USA). From each region of interest, mean standardized uptake values and target-to-background ratios were measured as indices of 18F-FDG uptake.Tissues were weighed and measured for 18F-FDG activity on a high-energy γ counter (Perkin Elmer). Uptake levels were expressed as percentage of injected dose per gram of tissue (%ID/g). Acquisition of no enhanced CT images was followed by emission PET imaging. Attenuation correction was not applied. Care was taken to maintain an environment of 23-25°C for the mice.
Immunofluorescence. CD31, phosphor-STAT3 (Tyr705), and VEGFR2 positive vessels and tumor areas were measured using a ZEN 710 confocal microscope program (Geneva, Switzerland) according to the manufacturer's instructions (https://www.zeiss.com/microscopy/int/products/microscope-software/zen-lite.html). CD31 antibody (Abcam, Cambridge, UK), phosphor-STAT3, VEGFR-2 (Santa Cruz Biotechnologies, Dallas, TX, USA) goat anti-rabbit Alexa Fluor 488 (IgG H&L), and goat anti-mouse Alexa Fluor 594 (IgG H&L) (Invitrogen, LifescienceTechnology, Grand Island, NY, USA) were applied. Positive vessels and tumor areas were counted and calculated in four fields at ×100 magnification.
Statistical analysis. All statistical analyses were performed using Graphpad Ver 5.0. (GraphPad Software, Inc, La Jolla, USA). The statistical significance of single comparisons was calculated using a Student's t test.
Results
17AAG reduced liposarcoma cell proliferation and migration. When we treated with the HSP90 inhibitor during 72 h, LPS 863 and 14GS-076 cells decreased their proliferation at the 400 nM of concentration of 17AAG from 24 h (Figure 1A and B). To confirm the optimal concentration for the cell viability and signaling pathway for growth, we treated 17AAG at a concentration from 0 to 500 nM for 48 h (Figure 2A and B). From the fluorescence result, we found the cell growth decreased significantly from mostly 100 nM dose of 17AAG on both of liposarcoma cells. Through these results, it was confirmed that the inhibitory effects were dependent on the dose of 17AAG and half of cells were died when we treated with dose of 400-500 nM for 48 h.
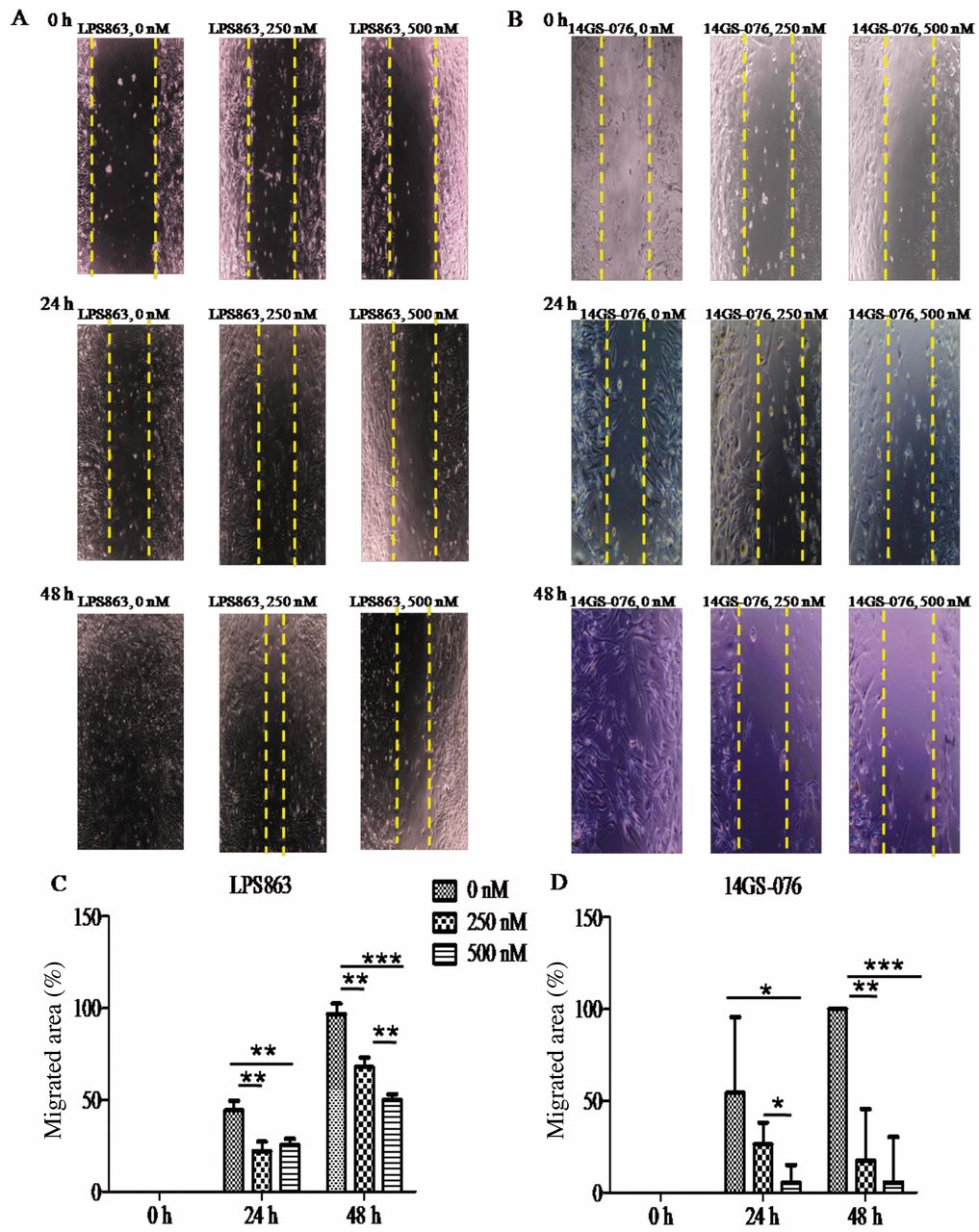
To determine the effect of HSP90 inhibitor on cell migration, we treated LPS863 and 14GS-076, liposarcoma cells with 17AAG (0-500 nM: 0-48 h). After drug treatment, we found the migration of LPS863 and 14GS-076 with 17AAG treated groupsreduced significantly than in the control (Figure 3A-D). These results demonstrated the 17AAG showed an inhibitory efficacy on both of cell motility, mobility.
17AAG reduced the oncogenic cascade: AKT, ERK and STAT3. Next, we determined the signaling pathways associated with the oncogenic cascade on liposarcoma cells according to treatment with the Hsp90 inhibitor. When we treated LPS863 and 14GS-076 cells with 17AAG, p-AKT, p-ERK, and p-STAT3 showed a tendency to diminish compared to control groups in both cell lines (Figure 4). We hypothesized that the inhibitory effect on cell growth by 17AAG is achieved through an inactivation of the AKT and ERK pathways. In addition, given the reduced expression of STAT3 following17AAG treatment, we suspected the HSP90 inhibitor, 17AAG could suppress growth of liposarcoma but also it could inhibit angiogenesis event.
HSP90 inhibitor 17AAG inhibits the angiogenic factors in liposarcoma cells. To determine the inhibitory effect of the HSP90 inhibitor on angiogenesis in the LPS863 and 14GS-076cells, we treated each cell line with 17AAG for 48 h at a concentration of 500 nM. After treatment, we examined the mRNA expression levels of IL-8, VEGF and IL-6 using qPCR. The results of real time PCR showed mRNA expression levels of angiogenic factors (VEGF and IL-8) were decreasing in all of the treated groups (Figure 5). Another agonistic factor for angiogenesis, IL-6, also decreased in LPS863 and 14GS-076 cells significantly. From these results could determine that HSP90 inhibitor could regulate transcription levels of angiogenesis factors on liposarcoma development.

Treatment with17AAG (500 nM) for 48 h. After applying the HSP90 inhibitor on LPS863 (A) and 14GS-076 (B), checked up the viability using Cell Tighter-Fluor™ mixture. These results confirmed the dose of 17AAG (500 nM) was optimal condition for following experiment. Data are shown relative to PBS-treated controls. *p<0.05, **p<0.005, ***p<0.0005.
Effect of HSP90 inhibitor on tumor growth in vivo. To evaluate the therapeutic effects of targeting HSP90 in vivo, we established a mouse model by subcutaneous injection of LPS863 into immune deficient mice. Administration of 17AAG was performed during 3-times a week over five weeks to evaluate tumor size (Figure 6A-E). After 5 weeks of treatment with 17AAG, we observed the tumor volume decreased significantly on this tumor bearing mice.

Effect of 17AAG, an HSP90 inhibitor on migration. LPS863 and 14GS-076 cells were treated with 17AAG (0, 250 and 500 nM: 0-48 h) for migration assay. Representative images of the control and treatment groups were obtained at the end of the experiment (A, B). 17AAG markedly diminished cell migration of LPS863 and 14GS-076cells, time- and dose-dependently compared to the control group (C, D). *p<0.05, ** p<0.005, *** p<0.0005.

Western blotting for cell viability. Oncogenic protein levels of phosphor-STAT3, phosphor-AKT, and phosphor-ERK were estimated using western blotting. 17AAG treatment (250 and 500 nM, for 48 h) led to decreased STAT3, AKT, and ERK activities in LPS863 and 14GS-076 cells.
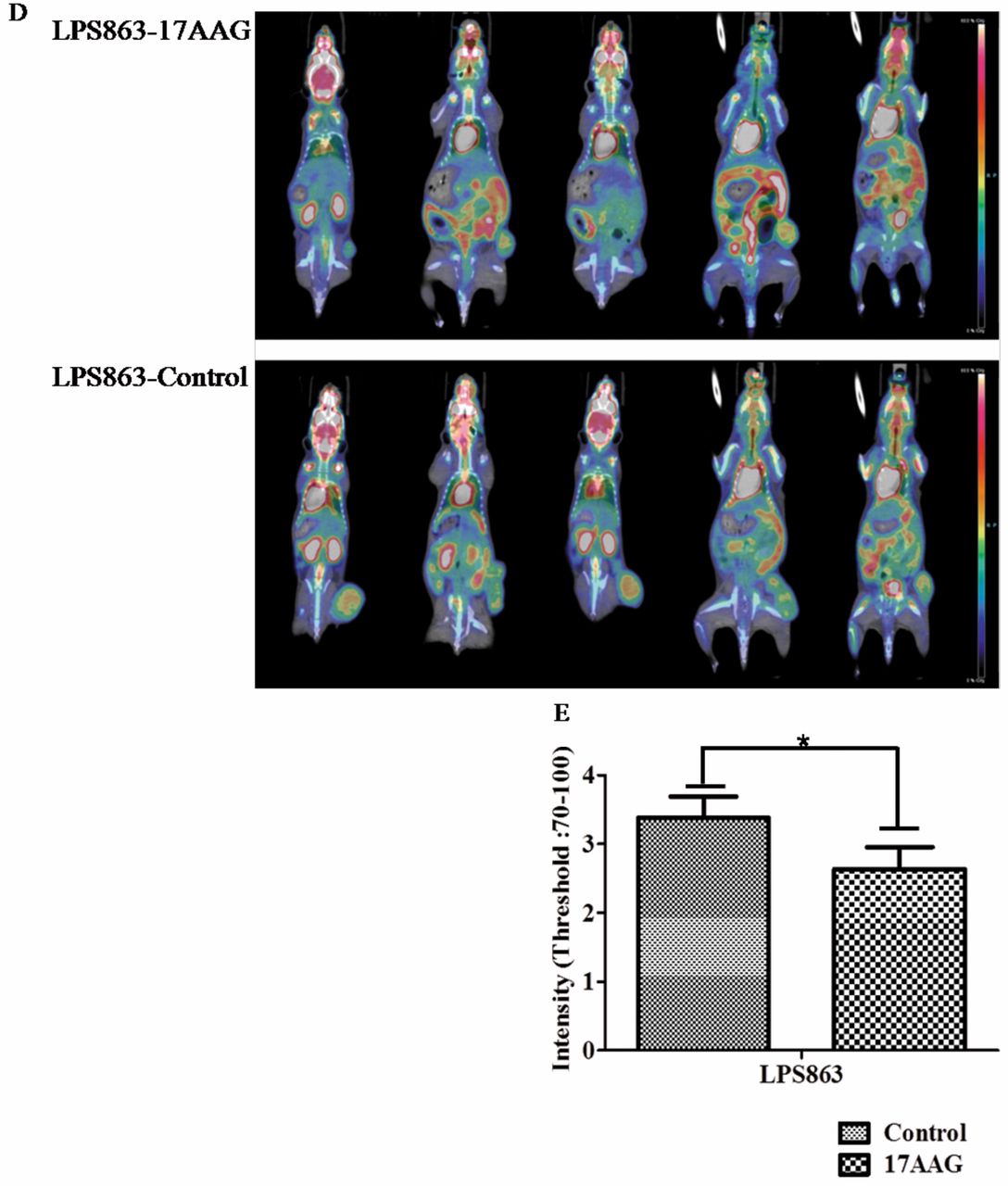
Therapeutic effects of HSP90 inhibitor on VEGFR2 of tumor tissues. From these results, we asked the main cause of efficacy of this drug, especially related to angiogenesis. To investigate the reasonsforthese anti-angiogenic effects, firstly, we tested for CD31, a marker of blood vessels, by tissue staining (20); as expected, we found a reduced expression levels of CD31 in the LPS863 model (Figure 7A). And about another crucial angiogenic factor, immuno-fluorescence expression levels of VEGFR2 also markedly reduced on the mouse tumor tissue (Figure 7B). Importantly, we also found the expression of activated STAT3 decreased after 17AAG treatment (Figure 7C). These results demonstrated that blocking of HSP90 reduced the main factors for angiogenesis and vascularizaion of liposarcoma (Figure 7D). These results showedthat 17AAG could inhibit not only tumor growth but also angiogenesis and vascularization of liposarcoma effectively in vivo system.
Discussion
HSP90 inhibition has been verified to affect cancer progression in various groups (16, 17). Liposarcoma is composed of various histological subtypes: well-differentiated dedifferentiated liposarcoma (WD/DDLPS), myxoidliposarcoma (MLPS) and pleomorphic liposarcoma (PLPS) (21). WD/DD LPS and MLPS show distinct differences in their molecular genetics (22, 23). WD/DDLPS has been shown to have MDM2 and CDK4 amplifications (22). Therefore, the cell-cycle progress induced by CDK4 amplification has been suggested to be an important factor of the disease. MLPS has a FUS-CHOP gene that results in translocation t(12;16) (q13;p11), and it occurs in 95% of cases (23). Receptor tyrosine kinase (RTK), MET, RET and PI3K signaling have been suggested as targetable pathways in myxoid liposarcoma (24). Because those treatments depend on the liposarcoma subtypes, it is necessary to establish therapeutic tools dependent on cell type. Recently, Steinmann et al. reported the significant cytotoxic effects in myxoid liposarcoma cell lines after treatment with the HSP90 inhibitor NVP-AUY922 (9). They also showed that the effect of this Hsp90 inhibitor was produced via the RAF/MEK/ERK and PI3K signaling pathways.

Detection of IL-8, IL-6, and VEGF expression levels in liposarcoma cells after treatment with 17AAG. LPS863 and 14GS-076 cells were treated with PBS (as controls) or with 17AAG (500 nM) for 48 h. RNA samples were isolated and subjected to real-time PCR analysis. Data were normalized to GAPDH levels and presented as fold changes in fluorescence density compared to that of the control group. Data are shown as mean±SD. *p<0.05 versus control.
Herein, we observed inhibitory effects on cell growth via inactivation of AKT and ERK when we treated patient-derived liposarcoma cells with 17AAG, an HSP90 inhibitor. We observed the anti-angiogenic effect of the HSP90 inhibitor in both in vitro and in vivo models that were similar to other studies except in p-ERK expressions in vivo (25). Recently, others studies showed the anti-cancer efficacy of Hsp90 inhibitor, also showed the decreasing effect on AKT and ERK expression levels not only in vitro, but in vivo (26, 27). Contrary to these data, in our mice models, it showed no differences in ERK expression levels in treated with 17 AAG compared to control groups (Figure 8). These results may attributed to the different cell backgrounds and microenvironments. Another reason may be that 17 AAG could not inhibit MEK-ERK pathway effectively in 25 mg/kg which was the dose used in our mice models. However, overwhelmingly, anti-cancer effect of this inhibitor of HSP90 was observed not only on proliferation and growth but also on angiogenesis in liposarcomas. Moser et al. showed that when they treated ECs and pericytes with an Hsp90 inhibitor, the activity of ERK and AKT induced by VEGF-A was decreased (25). Furthermore, the expression of VEGFR2 has been shown to decrease in ECs following treatment with the 17-DMAG HSP90 inhibitor. Recently, it has been stated that VEGFR2 may induce the activation of the PI3K-AKT pathway between endothelial cells and tumor cells through the Met receptor and integrin receptor RTK (28). It has also been argued that autocrine VEGF signaling has a major role in tumor formation (29, 30). Similar to findings by Lang et al., our data showed a significant difference in the mRNA levels of IL-8, IL-6, and VEGF between the control and 17AAG treated group in vitro (31). In agreement with the in vitro results, we observed that angiogenic factors significantly decreased in tumor tissues in our mouse model. Similar to the diminished VEGF mRNA expression in vitro, VEGFR expression levels were markedly decreased compared to control in vivo too.
STAT3 is constitutively activated in a variety of cancers. Furthermore, many tumor-produced factors, such as IL-6, IL-10 and VEGF, are activated by STAT3 both in tumor cells and the microenvironment as well as for the development of angiogenesis (32). IL-6 stimulates the activity of STAT3 in tumor cells, sequentially activating STAT3 as transcription factors trigger diverse oncogenic proteins (e.g., VEGF) and migration cascades (e.g., N-cadherin) (33). In the 17AAG-treated group of the LPS863 xenograft model, the positive area for p-STAT3 were diminished compared to the control group, and from these results, we thought that blockade of HSP90 could inhibit the crucial factors for angiogenesis in liposarcoma, such as STAT3 (34, 35). In keeping with these results, we suggest that inhibition of HSP90 can affect the tumor microenvironment for tumour growth such as angiogenesis. Finally, in the case of liposarcoma cell lines and xenograft models derived from LPS863 cell lines, we suggest that the therapeutic strategies for effective suppression of liposarcoma are PI3K-AKT pathway and the IL6-STAT3–VEGF pathway for cell growth and angiogenesis. Conclusively, this study showed that HSP90 inhibitor; 17AAG had antitumor efficacy against liposarcoma in vitro and in vivo. We suggest that 17AAG may have a potential anti-cancer drug for treating the liposarcoma patient.
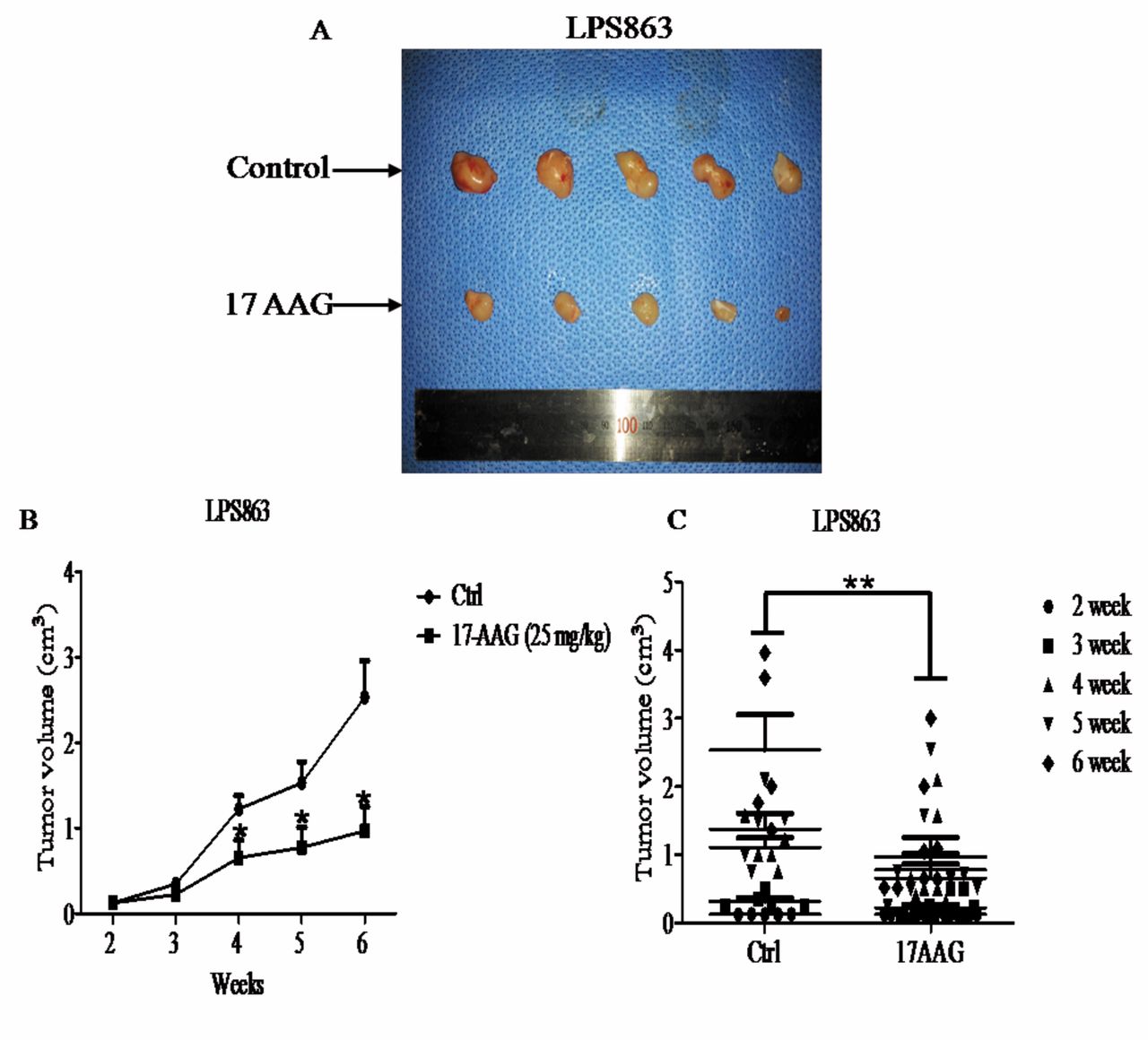

Impact of HSP90 inhibition on tumor growth of a xenograft mouse model. Liposarcoma LPS863 cells (2×107) were subcutaneously injected into the right flank of NSG mice. Two groups served as the control and treatment groups with 6-7 mice per group. When tumors reached a palpable size, the treated group was injected intraperitoneally with 25 mg/kg 17AAG three times per week. (A) Representative images of the control and treatment groups were obtained at the end of the experiment. (B, C) Tumor volume is shown in cm3 and was calculated as [length × length × height] ×0.5, *p=0.018, **p=0.0052. (D) MicroPET images of 18F-FDG uptake in the LPS863 cell line tumor-bearing mice at 6 week after treatment with 25 mg/kg of 17AAG. (E) The inhibitory effect of 17AAG markedly decreased tumor growth of the LPS863 cell line compared to the control group (*p<0.05 versus control).

Effects of HSP90 blockade on endothelial cells in an in vivo model. (A-C) After mice were treated with 17AAG (25 mg/kg), immunofluorescence staining for CD31, VEGFR2 and p-STAT3 in 17AAG xenograft tissue sections was performed. To measure fluorescence-positive vessel area, we used a ZEN 710 confocal microscope. (A, D) Densitometric analysis of the CD3-positive vessel area was down-regulated in the 17AAG-treatment group compared to control, which also indicated reduced angiogenesis in the 17AAG-treatment group of LPS863 (*p=0.0125). (B, D) HSP90 inhibition had a substantial effect on VEGFR2 expression in blood vessels on tumor sections from the LPS863 xenograft mouse model, ***p=0.0002. (C, D) Densitometric analysis of phosphor-STAT3 positive areas revealed a correlation with reduced angiogenesis in the 17AAG treated group of the LPS863 xenograft model; ***p=0.0002.

Effects of HSP90 blockade on tumor cells in vivo model. (A, B) After mice were treated with 17AAG (25 mg/kg), immuno-fluorescence staining was performed for p-AKT (Ser427), p-ERK (Thr732) in control and engrafted tumor tissue sections. HSP90 inhibitor (17AAG) effectively suppressed the expression level on p-AKT, *p<0.05 (A), however, expression levels of p-Erk (Thr732) were not diminished on the LPS863 tumor model (B).
Acknowledgements
This work was supported by grant of the Coopertative Research Program for Agriculture Science & Development (Project No. PJ 01100202) supported by rural Development Administration, Republic of Korea.
Footnotes
↵* These Authors contributed equally to this study.
- Received July 25, 2017.
- Revision received September 11, 2017.
- Accepted September 18, 2017.
- Copyright© 2017, International Institute of Anticancer Research (Dr. George J. Delinasios), All rights reserved
References
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- No citing articles found.