


Abstract
Background: Castration-resistant prostate cancer (CRPC)-related deaths are increasing worldwide. Therefore, clarification of the mechanisms of hormone-related tumor progression and resistance to anti-androgen drugs is useful in order to develop strategies for appropriate treatment of CRPC. Galectin-3 has been shown to be correlated with tumor progression in a variety of cancer types through the regulation of tumor proliferation, angiogenesis, and apoptosis. Materials and Methods: We examined tumor cell invasion and migration using the xCELLigence system. Control LNCaP and galectin-3-expressing LNCaP (LNCaP-Gal-3) cells were cultured with androgen-depleted medium with 5% charcoal-stripped serum. Cells were treated for 24 h with or without dihydrotestosterone alone or combined with MDV3100 and bicalutamide; gene profile was then analyzed by microarray analysis and mRNA expression was confirmed by quantitative real-time polymerase chain reaction (qRT-PCR). We evaluated tumor growth using spheroids and xenograft tumor growth in a mouse model. Results: In vitro, LNCaP-Gal-3 cells promoted both cell migration and invasion in an androgen-independent manner compared to control LNCaP cells. Galectin-3 also enhanced anchorage-independent growth and xenograft tumor growth even after castration. Importantly, galectin-3 greatly enhanced transcriptional activity of the androgen receptor (AR), especially on treatment with dihydrotestosterone. In microarray and qRT-PCR analyses, galectin-3 increased the expression of several AR-target genes, such as kallikrein-related peptidase 3 (KLK3), and transmembrane protease, serine 2 (TMPRSS2). These AR-target genes were not fully suppressed by anti-androgen drugs such as bicalutamide or MDV3100. Galectin-3 significantly inhibited the effect induced by anti-androgen drugs MDV3100 and bicalutamide, suggesting that galectin-3 may be involved in resistance to anti-androgen drug through enhancement of transcriptional activity of AR and expression of AR-related genes. Conclusion: These results suggest that galectin-3 is a potential target molecule for future treatment of anti-androgen drug-resistant prostate cancer.
Prostate cancer is one of the most common types of cancer among men worldwide and has recently become one of the leading causes of cancer-related death (1-3). The American Cancer Society's estimate for prostate cancer in the United States in 2015 was about 220,800 new cases per year in the US (4, 5). Although early-stage prostate cancer is well treated by surgical resection or radiotherapy, such as prostate brachytherapy and intensity-modulated radiation therapy, advanced-stage prostate cancer, especially castration-resistant prostate cancer (CRPC), has limited therapeutic options, based mainly on hormone therapy.
Galectins are members of the animal lectins, which are a family of carbohydrate-binding proteins characterized by their affinity for β-galactoside and by a conserved sequence of the carbohydrate recognition domain (6, 7). Galectins are mainly found in the cytosol and the nucleus, and 14 members have been identified. Galectins are classified into three subgroups based on their structure, including: (a) prototype (galectin-1, -2, -5, -7, -10, -11, -13, and -14); (b) chimera type (galectin-3); and (c) tandem repeat type (galectin-6, -8, -9, and -12) (7). Galectin-3 expression is increased in a variety of neoplastic cell types and has been associated with many tumor development processes including cell growth, adhesion, proliferation, and metastasis (7). Moreover, clinical evidence suggests that there is a correlation between galectin-3 expression and malignant potential in some types of tumors, such as colon cancer and thyroid cancer. Although there have been many reports that galectin-3 increases the aggression of several kinds of cancer, there are relatively few reports concerning the role or galectin-3 roles in prostate cancer (8, 9). We previously reported that cytoplasmic galectin-3 was strongly implicated in anti-apoptotic function in prostate cancer that was induced by an anticancer drug through an effect on the stability of mitochondria (10).
The effects of galectin-3 on prostate cancer cell migration, invasion, anchorage-independent growth, and especially on resistance to anti-androgen drugs, were investigated here.
Materials and Methods
Cell culture. The prostate cancer androgen-dependent cell line LNCaP and LNCaP cells transfected with Gal-3 (LNCaP-Gal-3) (10, 11), and the metastatic type of the androgen-independent cell lines DU145 and PC3 that were used in this study were obtained from the American Type Culture Collection (ATCC; Manassas, VA, USA). All cells were maintained in RPMI-1640 medium containing 2 mmol/l glutamine, penicillin, and streptomycin (Life Technologies, Carlsbad, CA, USA) and 10% (v/v) fetal bovine serum in 5% CO2 at 37°C using standard cell culture procedures (10). At 24 h before treatment with the anti-androgen drug MDV3100 (Santa Cruz Biotechnology, Dallas, TX, USA) or with bicalutamide (Wako Pure Chemical, Osaka, Japan), the cells were transferred to RPMI-1640 phenol red-free medium for 24 h. 10 μM MDV3100 and 10 μM bicalutamide were dissolved in dimethyl sulfoxide (Kanto Chemical Co, Inc, Tokyo, Japan) and 1 nM dihydrotestosterone (DHT) (Sigma Aldrich, St. Louis, MO, USA) in methanol.
Primary antibodies. The primary antibodies used in this study were: anti-androgen receptor (AR) (clone number AR441i DAKO North America, Inc. Carpinteria, CA, USA) and TIB166 monoclonal rat anti-gal-3 from the ATCC (10, 11), and anti-human β-actin antibodies (Clone number A5441); (Sigma-Aldrich).
Real-time quantitative polymerase chain reaction (qPCR). Total RNA was extracted from cultured cells using the RNeasy micro kit (Qiagen, Hilden, Germany), as previously described (12). The appropriate dilutions of each single-stranded cDNA were prepared for subsequent PCR amplification, and reactions using β-actin were monitored as a quantitative control. Each sample was analyzed in triplicate for each primer pair. The relative expression levels of prostate-specific antigen (PSA), transmembrane protease, serine 2 (TMPRSS2), and galectin-3 (LGALS3) normalized to β-actin (ACTB) were calculated using the relative standard curve method and Light Cycler Software Ver.3.5 (Roche Diagnostics, Madison, WI, USA). The primer sequences were as follows: PSA: F-GCATGGGATGGGGATGAAGTAAG, R-CATCAAATCTGAGGGTTGTCTGGA; TMPRSS2: F-CCATTTGCAGGATCTGTCTG, R-GGATGTGTCTTGGGGAGCAA; LGALS3: F-GCCTTCCACTTTAACCCACG, R-ACTGCAACCTTGAAGTGGTCA; and ACTB: F-GAGGTGATAGCATTGCTTTCG, R-CAAGTCAGTGTACAGGTAAGC.
Western blot analysis. Cell lysates were prepared using the Mammalian Cell Extraction kit (Bio Vision, Mountain View, CA, USA). After centrifugation at 180 × g for 10 min, the supernatant was collected as the total protein extract and stored at −80°C. Protein concentration was then measured using a protein assay reagent (Bio-Rad Laboratories, Hercules, CA, USA). Equal amounts of protein were separated by sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis and electrophoretically transferred to a polyvinylidene difluoride membrane. The membrane was then blocked with Block Ace (Dainippon Pharmaceutical, Osaka, Japan) for 60 min and incubated with primary antibody at 4°C overnight. Blots were developed by chemiluminescence using a horseradish peroxidase-coupled secondary antibody (Invitrogen) and the ECL-Plus Kit (GE Healthcare, Piscataway, NJ, USA) (10).
Cell migration and invasion assays. Invasion and migration assays of LNCaP with and without galectin-3 expression were performed using the xCELLigence RTCA Analyzer (Roche Applied Science, Mannheim, Germany) in a humidified atmosphere of 5% CO2 and 95% air at 37°C (13). The medium of cells in a cell invasion and migration (CIM) plate (ACEA Biosciences, San Diego, CA, USA) containing cells at a density of 4×104 cells per well in transfection solution was replaced with fresh RPMI 1640 phenol red-free medium supplemented with 5% FBS. The lower well was filled with 160 μl of phenol red-free with 10% FBS. The invasion assay was tested on a tumor invasion system in Matrigel (BD Biosciences, Bedford, MA, USA), diluted 1:40 with phenol red-free medium (as detailed by Roche in the application notes) coated onto 16-well plates. Cells were further incubated for measurement of changes in the cell index over time intervals between 0 to 60 h. The cell index shows cell invasion or migration status based on the measured electrical impedance.
Luciferase assay. LNCaP and LNCaP-Gal-3 cells were seeded in 12-well plates (2×105 cells/well) after counting. After 24 h, the medium was changed to phenol red-free RPMI-1640 with 5% charcoal-stripped FBS. Cells were then co-transfected with pGL4.74 firefly luciferase reporter plasmid and Renilla luciferase PSA plasmid (14) using Lipofectamine® 2000 (Invitrogen). After 24 h of transfection, the medium was changed to phenol red-free RPMI-1640 supplemented with DHT and MDV3100 for 24 h. Subsequently, cell lysates were prepared, and luciferase activity was measured using the Dual-Luciferase Reporter Assay system (Promega, Madison, WI, USA). Firefly luciferase activity was normalized to Renilla luciferase activity (15, 16).
Spheroid production. Spheroids were prepared from single-cell suspensions of prostate cancer cell lines, as previously described (17-19). In brief, cells at a density of 0.5×104 cells per ml in RPMI-1640 phenol red-free medium supplemented with 5% (v/v) charcoal-stripped FBS were placed in the wells of a 96-well ultra-low attachment (ULA) plate (17-19). The ULA plate was then centrifuged at 100 × g for 3 min at room temperature. After 72-h incubation, when spheroids had formed, the medium was exchanged with fresh RPMI phenol red-free medium supplemented with DHT and MDV3100 (150 μl/well) followed by medium exchange every 48 h with care not to disturb the spheroids. Spheroids were cultured for 7 days before final analysis (20, 21). Images of all spheroids were taken daily for growth determination and cytotoxicity experiments using an Olympus IX53 microscope with a ×10 objective and an attached Olympus DP22/27 (Olympus Corporation, Tokyo, Japan) camera. Spheroid size was measured by open-source software ImageJ (ImageJ, version 1.48; NIH, Bethesda, MD, USA). We obtained descriptive data by Microsoft Excel (Microsoft Corporation, Redmond, WA, USA). and statistical analysis was performed by StatView 5.0 (SAS Institute, Inc., Cary, NC, USA).
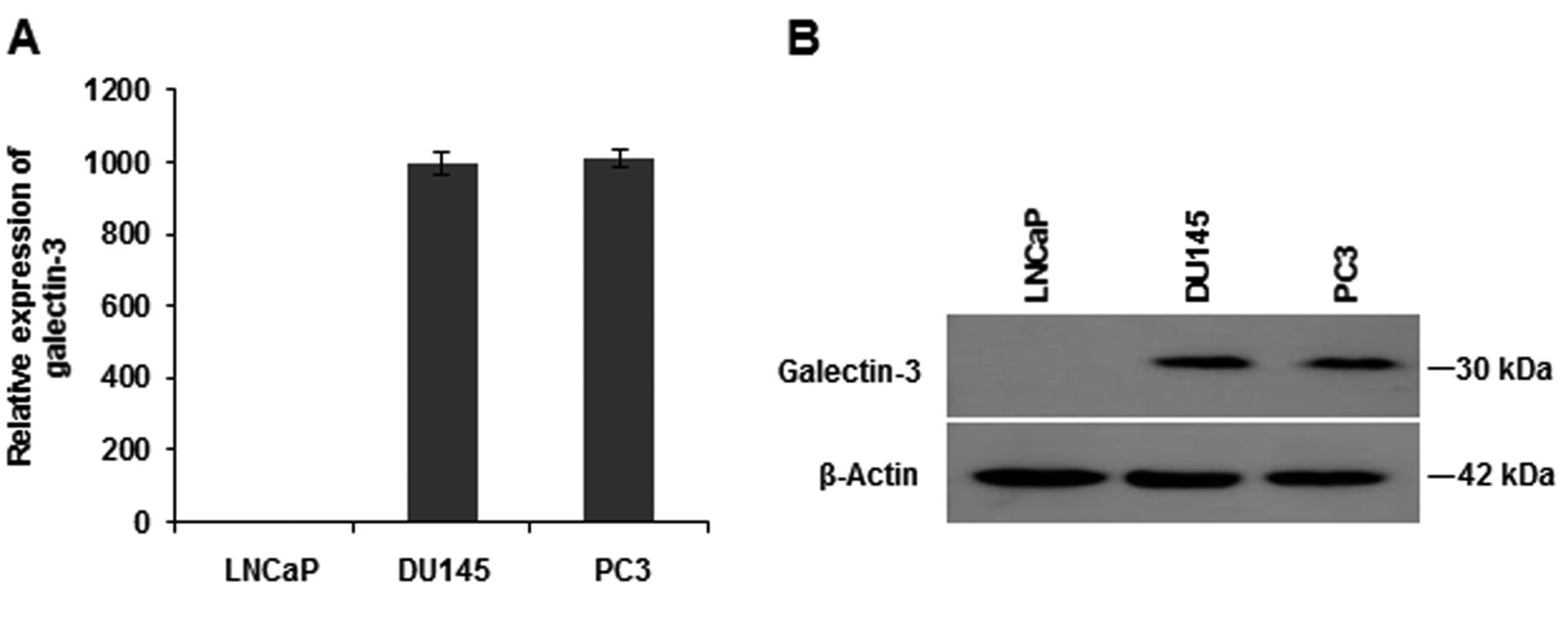
Endogenous galectin-3 expression in prostate cancer cell lines. Endogenous expressions of galectin-3 mRNA and protein in the androgen-dependent LNCaP and the androgen-independent DU145 and PC3 cell lines were examined using quantitative real-time polymerase chain reaction (A) and western blot analysis (B). Data in (A) are the means±SE.
Mouse xenograft model. BALB/c nude mice that lack a thymus and are unable to produce T-cells were used (Charles River Laboratory, Wilmington, DE, USA). LNCaP and LNCaP-Gal3 cells (8×106) suspended in 200 μl of Dulbecco's phosphate-buffered saline with 50% Matrigel (BD Biosciences) were subcutaneously inoculated into the right flank of all mice. We then divided mice into four groups: LNCaP and LNCaP-gal-3 for non-castrated groups, LNCaP and LNCaP-gal-3 for castrated groups. When each xenograft tumor reached a volume of approximately 200 mm3, the mice of the castrated groups were castrated by orchiectomy. Further growth of the tumors was monitored in all groups (22-24).
Microarray analysis. Total RNA from LNCaP and LNCaP-Gal-3 cells exposed to DHT, MDV3100, or bicalutamide for 24 h was extracted using an RNeasy Mini Kit (Qiagen) according to the manufacturer's instructions. RNA was quantified using a NanoDrop-1000 spectrophotometer, and quality was monitored with an Agilent 2100 Bioanalyzer (G2938C; Agilent Technologies, Palo Alto, CA, USA). Cyanine-3 (Cy3)-labeled cRNA was prepared from 0.2 μg RNA using the Low Input Quick Amp One-Color Labeling Kit (Agilent) according to the manufacturer's instructions. Dye incorporation and cRNA yield were checked with a NanoDrop ND-1000 spectrophotometer. Cy3-labelled cRNA (specific activity >15.0 pmol Cy3/μg cRNA) was fragmented at 60°C for 30 min and hybridized to Agilent SurePrint G3 Human GE Oligo Microarrays Ver3.0 (G4858A#072363) for 17 h at 65°C in a rotating Agilent hybridization oven. After hybridization, the microarrays were washed for 1 minute at room temperature with GE Wash Buffer 1 (Agilent) and for 1 min at 37°C with GE Wash buffer 2 (Agilent). After washing, slides were scanned immediately on an Agilent DNA Microarray Scanner (G2505C) using a one-color scan setting for 8×60 k array slides (scan area 61 mm × 21.6 mm, scan resolution 3 μm, dye channel set to green). The scanned images were analyzed with Feature Extraction Software 10.7 (Agilent) using default parameters (protocol GE1-107_Sep09 and Grid: 072363_D_F_20150612). The microarray data across all chips and genes were analyzed using GeneSpring (version 13.0) and normalized by 75 percentile shift (25, 26).
Statistical analysis. All statistical analyses were performed using StatView 5.0 (SAS Institute, Inc., Cary, NC, USA). Parameters were compared by Student's t-test, and ANOVA was used to compare the groups with more than two parameters. A difference of p<0.05 was considered significant.
Results
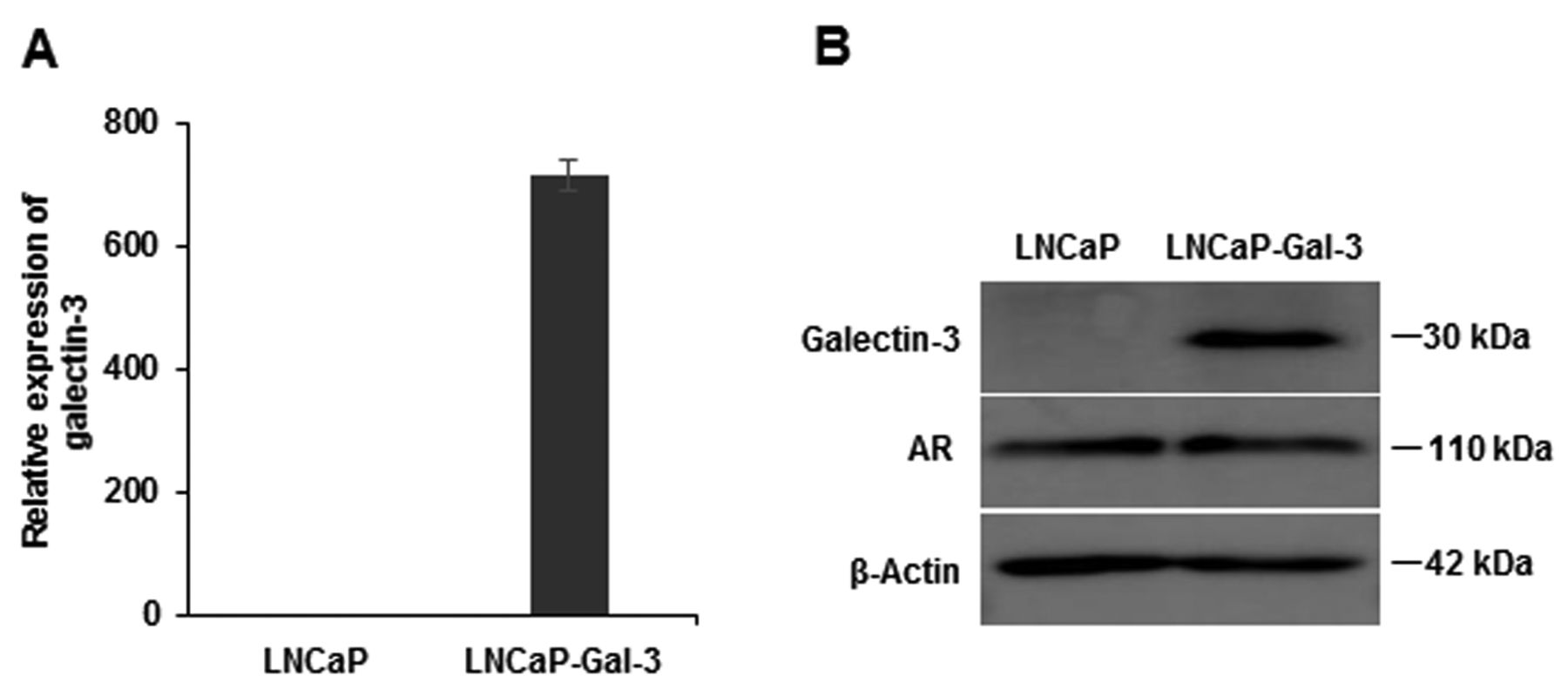
Galectin-3 expression in prostate cancer cell lines. The mRNA and protein expression levels of endogenous galectin-3 were measured in several prostate cancer cell lines using qRT-PCR and western blotting, respectively. AR-expressing LNCaP cells showed no expression of endogenous galectin-3, but non-AR-expressing DU145 and PC3 cells showed strong expression of galectin-3 on both qRT-PCR and western blotting assays (Figure 1). These results suggest that androgen-dependent LNCaP cells do not express galectin-3, whereas androgen-independent cells (DU145 and PC3) highly express galectin-3. LNCaP cells stably-expressing galectin-3 (LNCaP-Gal-3) were also used in this study. As expected, these cells highly expressed galectin-3 on RT-PCR and western blotting assays; there was no difference in AR expression between LNCaP-Gal-3 cells and LNCaP cells on qRT-PCR and western blotting (Figure 2).
Effects of galectin-3 on migration and invasion. The effects of galectin-3 expression on cell migration and invasion abilities were measured using the xCELLigence system. LNCaP-Gal-3 cells showed significantly enhanced migration compared to control LNCaP cells (p=0.0005) (Figure 3, upper panels). Cellular invasion was also increased in LNCaP-Gal-3 cells compared to control LNCaP cells (p=0.0002) (Figure 3, lower panels). The migration and invasive abilities of these cells were not enhanced by DHT treatment (Figure 3, right), suggesting that there might be an AR-independent pathway in galectin-3-induced cell migration and invasion. These results clearly show that galectin-3 significantly promotes cell migration and invasion in LNCaP cells, which suggests that galectin-3 may be involved in tumor invasion and metastasis in an androgen-independent manner.
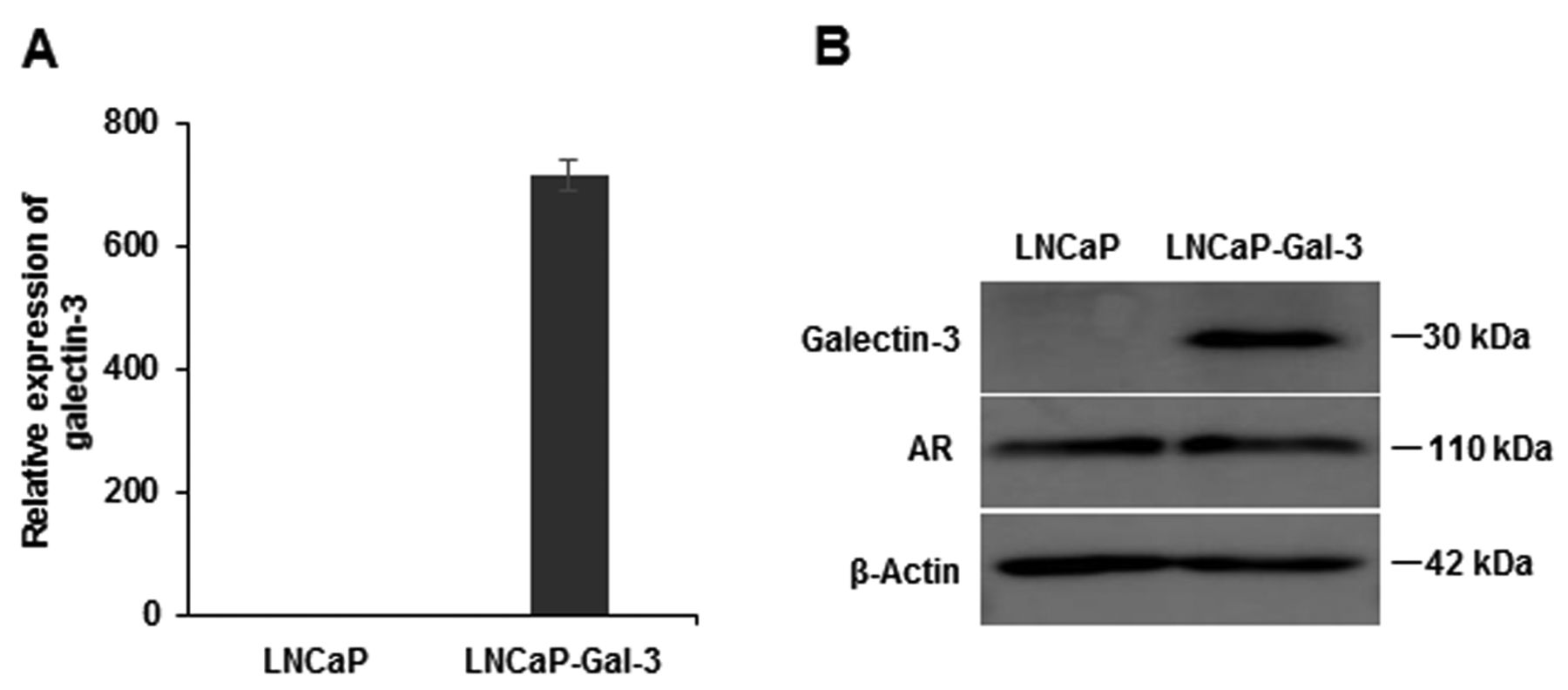
Galectin-3 and androgen receptor (AR) expressions in galectin-3 expressing LNCaP cells (LNCaP-Gal-3). The expressions of galectin-3, AR, and β-actin in LNCaP-Gal-3 cells were analyzed by western blot analysis (A), and galectin-3 expression was analyzed not only by quantitative real-time polymerase chain reaction (A), but also western blot (B). Data in (A) are the means±SE.
Galectin-3 enhances anchorage-independent growth of LNCaP cells. To evaluate the effect of galectin-3 on the anchorage-independent growth ability of the cells, 3D spheroid growth assays were performed using both LNCaP-Gal-3 and control LNCaP cells, with and without DHT treatment and in the presence or absence of the anti-androgen agent MDV3100. DHT treatment greatly increased spheroid formation of LNCaP-Gal-3 cells compared to control LNCaP cells (p=0.01) (Figure 4). These results indicate that galectin-3 enhances anchorage-independent growth, especially with DHT treatment.
Galectin-3 enhanced the expression of AR-target molecules such as KLK3 and TMPRSS2. To examine the genes involved in the tumor growth enhancement by DHT treatment and galectin-3, the expression of 41 AR-target genes after DHT treatment with/without anti-androgen drug treatment (bicalutamide or MDV3100) were compared in control LNCaP and in LNCaP-Gal-3 cells by microarray analysis (Figure 5). AR inhibition by MDV3100 or bicalutamide treatment before DHT treatment of LNCaP cells attenuated expression of an androgen-induced gene set. On the other hand, LNCaP-Gal-3 cells treated with DHT overexpressed AR-target molecules such as KLK3 or TMPRSS2 (Figure 5), and the expression levels of these molecules were not fully inhibited by MDV3100 or bicalutamide, supporting the mechanism of anti-androgen drug resistance by galectin-3 (Figure 5).
Galectin-3 suppresses anti-androgen drug effects. To evaluate the effects of galectin-3 on resistance to anti-androgen drug in prostate cancer cells, the mRNA levels of PSA and TMPRSS2 were measured in DHT-treated LNCaP-Gal-3 and control LNCaP cells in the presence or absence of the anti-androgen agents MDV3100 or bicalutamide (Figure 6A and B). DHT treatment of LNCaP-Gal-3 cells significantly increased KLK3 and TMPRSS2 expression compared to control LNCaP cells. However, neither MDV3100 nor bicalutamide treatment significantly reduced KLK3 and TMPRSS2 expression even after a DHT-induced increase, suggesting that galectin-3 expression might be involved in resistance to anti-androgen drug in prostate cancer. A luciferase activity assay for PSA indicated that galectin-3 enhanced PSA transcriptional activity, especially in the presence of DHT (Figure 6C). Importantly, PSA transcriptional activity of LNCaP-Gal-3 cells after DHT stimulation was not fully inhibited by MDV3100, whereas that of control LNCaP cells was completely inhibited by MDV3100 (Figure 6C). These results combined suggest that galectin-3 has suppressive effects against anti-androgen treatment agents such as MDV3100 and bicalutamide, which are mediated through enhancement of PSA transcriptional activity.

Effects of galectin-3 expression in LNCaP-Gal-3 cells on cell invasion and migration with/without dihydrotestosterone (DHT). Migration over 60 h (A) and invasion as cell index at 48 h (B) of LNCaP and LNCaP-Gal-3 cells were assayed using the xCELLigence system. LNCaP-Gal-3 cells showed significantly enhanced cell migration and invasion compared to control LNCaP cells (p=0.0005 and p=0.0002, respectively). Significantly different at *p<0.05, and **p<0.01; NS: Not significantly different. Data are the mean, error bars indicate SD.
Galectin-3 enhances the growth of LNCaP xenograft tumors in an androgen-independent manner in BALB/c nude mice. The effect of galectin-3 on the growth of LNCaP xenograft tumors in BALB/c nude mice was examined next. When xenograft tumors reached a volume of approximately 200 mm3 after 4 weeks, mice with such tumors were castrated, and further growth of the tumors was monitored. The size of galectin-3-null control LNCaP xenografts shrank from more than 200 mm3 to 100 mm3 two weeks after castration (p=0.027). In contrast, LNCaP-Gal-3 xenograft growth increased even after castration. These results clearly show that galectin-3 inhibited tumor shrinkage induced by hormone deprivation therapy in this mouse model (Figure 7).
Discussion
Prostate cancer has become a common disease worldwide and is one of the leading causes of cancer-related death among men (27-29). Metastatic prostate cancer is commonly treated with hormone therapy, following which most cases eventually become resistant to androgen-deprivation therapy, the so-called CRPCs. These prostate cancer cells are heterogeneous, which is why several prostate cancer cell lines have different levels of aggression and metastatic ability. We found that DU145 and PC3, which display more aggressive phenotypes, expressed a high amount of galectin-3. Additionally, in terms of hormone sensitivity, hormone-sensitive LNCaP cells did not express galectin-3, in contrast to the hormone-insensitive DU145 and PC3 cells, which displayed both high mRNA and protein expressions of galectin-3 (Figure 1). In this study, we up-regulated galectin-3 in LNCaP cells and investigated its effects on the biochemical behavior of LNCaP cells.

Effect of dihydrotestosterone (DHT) stimulation of galectin-3-expressing LNCaP cells on spheroid formation. A: Spheroid sizes were measured on the sixth day after spheroid formation in different groups. B: Phase-contrast images of representative spheroids formed in the indicated cell lines with and without DHT stimulation. DHT stimulation of LNCaP-Gal-3 cells significantly enhanced spheroid formation compared to control LNCaP cells. *Significantly different at p<0.05.
Our data showed that galectin-3 overexpression in LNCaP cells contributed to increased cell growth, invasion, and migration in a DHT-independent manner, suggesting the association of galectin-3 with tumor invasion and metastatic events. These data supported previous findings that galectin-3 knockdown in PC3 cells reduced cell invasion and migration (6).
Generally, prostate cancers are androgen-dependent, and androgen deprivation therapies play a major role in therapeutic tactics as hormone therapy. Therefore, investigation of the mechanism of CRPC is important. A number of potential mechanisms of CRPC have been reported, including: i) AR overexpression; ii) promiscuous binding and activation of mutant AR by alternative ligands, such as estrogen, progesterone, glucocorticoids, bicalutamide, and flutamide; iii) ligand-independent mechanisms of AR activation via crosstalk with serine/threonine kinase 1 (AKT), erb-b2 receptor tyrosine kinase 2 (HER2), and AKT serine/threonine kinase 1 (AKT1) kinases that phosphorylate the AR, and via long non-coding RNAs that bind to the AR to stimulate transcription of AR target genes; and iv) AR-independent pathways through signal transducer and activator of transcription 3 (STAT3) signaling or up-regulation of anti-apoptotic BCL2, apoptosis regulator (BCL2) (30-34). In the present study, we revealed that galectin-3 activated PSA transcription and enhanced expression of AR target genes such as KLK3 and TMPRSS2. Moreover, we showed that treatment of LNCaP-Gal-3 cells with MDV3100 or bicalutamide did not fully suppress the expression of PSA and TMPRSS2, whereas their expression in LNCaP cells was completely suppressed by treatment with MDV3100 and bicalutamide, suggesting that galectin-3 might play important roles in resistance to anti-androgen drug through the regulation of the AR signal pathway.

Microarray analysis of control LNCaP and LNCaP-Gal-3 cells. Microarray analysis of overall gene expression in DHT- and drug-treated LNCaP and LNCaP-Gal-3 cells treated as indicated. Androgen receptor (AR)-target gene expression after androgen stimulation with/without anti-androgen drug treatment such as bicalutamide or MDV3100. The 41 AR target gene set in LNCaP-Gal-3 cells treated with DHT plus bicalutamide or MDV3100 compared with control LNCaP cells. LNCaP-Gal-3 treated with DHT overexpressed AR target molecules such as kallikrein-related peptidase 3 (KLK3) or transmembrane protease, serine 2 (TMPRSS2), and the expression levels of these genes were not fully inhibited by bicalutamide or MDV3100.

Relative expression of prostate-specific antigen (PSA) and transmembrane protease, serine 2 (TMPRSS2) in dihydrotestosterone (DHT)-treated LNCaP-Gal-3 and control LNCaP cells in the presence or absence of the anti-androgen agents MDV3100 or bicalutamide. Real-time polymerase chain reaction analysis of PSA (A) and TMPRSS2 (B) expressions in LNCaP and LNCaP-Gal-3 cells cultured in androgen-depleted media with 5% charcoal-stripped serum. The cells were treated for 24 h with or without DHT (1 nM) combined with mock, MDV3100 (10 μM), or bicalutamide (10 μM) and MDV3100 (10 μM). C: PSA transcriptional activity with and without galectin-3. Luciferase activity reporter assay of LNCaP and LNCaP-Gal-3 cells for PSA in the presence or absence of DHT treatment was performed. The luciferase reporter plasmid, pGL3PSAp-5.8, driven by a 5.8 kb PSA promoter, was used. DHT treatment was performed 24 h after transfection, and luciferase activity was quantified using a luminometer 48 h after transfection. Galectin-3 enhanced PSA transcriptional activity, especially in the presence of DHT. PSA transcriptional activity of LNCaP-Gal-3 cells after DHT treatment was not fully inhibited by MDV3100, because PSA transcriptional activity of LNCaP-Gal-3 cells treated with MDV3100 was still significantly higher than that of non-treated LNCaP-Gal-3 cells (C). **Significantly different at p<0.01; NS: not significant. Data are the mean, error bars indicate SD.
In contrast to LNCaP cells, overexpression of AR-target molecules such as NK3 homeobox 1 (NKX3-1) as well as TMPRSS2 and KLK3 in LNCaP-Gal-3 cells was still observed in the microarray even after treatment of the cells with MDV3100 or bicalutamide (Figure 5). Up-regulation of NKX3-1 supports the notion that galectin-3 might stimulate AR target molecules because NKX3-1 was found to collaborate with AR and regulate the AR transcriptional network, resulting in the promotion of prostate cancer cell survival (35).
Our results show that galectin-3 has an important role in tumor aggressiveness through the regulation of anchorage-independent cell growth, invasion, migration, and AR signaling. To the best of our knowledge, this is the first report to show that galectin-3 regulates AR signaling and is involved in resistance to anti-AR drugs. Furthermore, galectin-3 is implicated in anti-apoptotic function induced by hormone deprivation therapy in the mouse model, suggesting that galectin-3 has an important role in the tumor progression of CRPC. Galectin-3 is, therefore, considered to be a potential target molecule for the future treatment of CRPC.

Galectin-3 enhances the growth of LNCaP xenograft tumors in an androgen-independent manner in BALB/c nude mice. LNCaP and LNCaP-Gal-3 xenograft growth was measured once the tumors reached a size of 200 mm3, following castration or not. LNCaP xenografts shrank two weeks after castration (p<0.027), whereas LNCaP-Gal-3 xenograft growth was enhanced even after castration. **Significantly different at p<0.01 by 2-way ANOVA.
Acknowledgements
The Authors wish to thank Yoshiko Shimada and Aya Shinomiya for their assistance with experiments and Hideaki Horikawa (Tokushima University Graduate School, Support center for Advanced Medical Science) for technical support in microarray analysis. The Authors would like to express special thanks to Dr. Atsushi Mizokami (Department of Integrative Cancer Therapy and Urology, Kanazawa University Graduate School of Medical Science) for providing luciferase reporter plasmid, and Dr. Avraham Raz (Departments of Oncology and Pathology, Karmanos Cancer Institute, Wayne State University) for his helpful advice.
Footnotes
This article is freely accessible online.
Conflicts of Interest
The Authors declare no conflicts of interest.
- Received November 5, 2016.
- Revision received November 25, 2016.
- Accepted November 29, 2016.
- Copyright© 2017 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}