


Abstract
Background/Aim: Cholangiocarcinoma (CCA) is an aggressive cancer for which standard treatments are still ineffective. This study demonstrated the antiproliferative and anti-metastatic activity of metformin, an anti-diabetic drug, in CCA cells. Materials and Methods: Cell proliferation, migration/invasion and anoikis resistance were determined. The underlying mechanisms were identified using western blotting and immunocytofluorescence. Results: Metformin significantly suppressed proliferation of CCA cells in a dose- and time-dependent manner, regardless of glucose present in the medium. A low dose of metformin significantly increased anoikis and inhibited migration/ invasion of CCA cells that was in concert with the decrease of vimentin, matrix metalloproteinase (MMP)-2 and -7. Activation of 5’ adenosine monophosphate-activated protein kinase (AMPK) by phosphorylation together with suppression of nuclear translocation of signal transducer and activator of transcription 3 (STAT3) and nuclear factor-kappa B (NF-ĸB) were the underlying mechanisms for these effects. Conclusion: Metformin is a potent antiproliferative and anti-metastatic agent against human CCA cells. These findings encourage the repurposing of metformin in clinical trials to improve CCA treatment.
Cholangiocarcinoma (CCA) is the most common type of liver cancer after hepatocellular carcinoma worldwide. The incidence of CCA varies geographically: It is considered low in Western countries, but it is relatively high in the East and Southeast Asia. A high incidence of CCA has been noted in the Northeast of Thailand and infection with the liver fluke, Opisthorchis viverrini has been shown to be a strong risk factor for CCA in this region (1). Primary sclerosing cholangitis, hepatolithiasis, and hepatitis B virus infection, however, are the documented risk factors for CCA in Western countries (2, 3). CCA is a lethal type of cancer since early diagnosis is rarely possible. Most patients present at an inoperable advanced/metastatic stage. Chemotherapeutic and radiological therapies are alternative choices, but these treatments still have a low potential for curative purposes (4). The search goes on for additive treatments that might improve therapeutic outcome.
Development of a new pharmaceutical requires extensive research and development costs and time. Repositioning a drug that is already clinically used provides a way to shortcut the research and time required, and costs much less (5). Diabetes mellitus (DM) has been reported to be associated with risk and prognosis of many cancer types, including CCA (6, 7). The molecules that bridge DM and cancer have been shown to be insulin and a high blood glucose level (8-10). Repositioning an antidiabetic compound that appears to have great potential for cancer prevention and as a cure provides a rare opportunity. Metformin is such a drug.
Metformin is a widely studied drug for its potential for cancer treatment even though it is primarily used as a first-line treatment for type 2 DM. Metformin indirectly activates AMP-activated protein kinase (AMPK) by inhibiting the activity of mitochondrial complex I, resulting in an increased cellular AMP/ATP ratio. Activated AMPK plays an important role as a cellular energy sensor by promoting glycolysis and inhibiting gluconeogenesis. Apart from its function in energy metabolism, AMPK is able to interact with several signaling pathways and transcription factors such as phosphoinositide-3-phosphate/protein kinase B (PI3K/AKT), Janus kinase/signal transducer and activator of transcription (JAK/STAT), nuclear factor-kappa B (NF-ĸB) and the mammalian target of rapamycin (mTOR). The attempt to repurpose a drug for CCA, in this instance metformin, is a potential way to find an improvement for CCA treatment. An epidemiological study in the United States showed that metformin significantly reduced risk of CCA in patients with DM (11). The potential for repurposing metformin for cancer treatment has been reviewed (5). To date, 243 clinical trials of metformin as cancer treatment have been registered at https://www.clinicaltrials.gov. Metformin, however, is still at the preclinical stage for some cancer types, including CCA (12, 13).
In the present study, the effects of metformin on the proliferation and progression of liver fluke-associated CCA cells were examined. The effects of metformin on the metastatic potential, namely migration, invasion and anoikis resistance of CCA cells are reported. The molecular mechanisms by which metformin affected CCA cells were elucidated. The demonstrated effects of metformin on CCA cells in the present study should further encourage clinical trials to improve the treatment of CCA.
Materials and Methods
Chemicals and antibodies. Metformin (1, 1-dimethylbiguanide hydrochloride) was purchased from Sigma-Aldrich (St. Louis City, MO, USA) and dissolved in sterile distilled water. Primary antibodies to p-AMPKα (T183/172), AMPK, p-RB (S249/T252), Rb, Cyclin D1, MMP2, MMP7, NF-ĸB (p65) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA), anti-p21 was from Cell Signaling, (Danvers, MA, USA), anti-vimentin from Abcam (Cambridge, UK), and anti-actin from Sigma. The secondary antibodies for western blots were goat anti-mouse and donkey anti-rabbit from GE Healthcare (Buckinghamshire, UK), and rabbit anti-goat from Dako (Carpinteria, CA, USA). The secondary antibodies for the immunocytofluorescent study were fluorescein isothyocyanate (FITC)-conjugated goat anti-rabbit (Santa Cruz Biotechnology) and Alexa Fluor 568-conjugated goat anti-rabbit (Invitrogen, Eugene, OR, USA). Hoechst 33342 was purchased from Invitrogen.
Cell lines. CCA cell lines, namely KKU-055, KKU-100, KKU-213 and KKU-214, were originally established from primary cultures of liver fluke associated CCA tissues from Thai patients and obtained from the Japanese Collection of Research Bioresources Cell Bank, Osaka, Japan. Cells were maintained in Dulbecco's modified Eagle's medium (DMEM)-high glucose (HG; 25 mM) or normal glucose (NG; 5.6 mM) supplemented with 10% fetal bovine serum and 1% antibiotic-antimycotic (all from Invitrogen). Cells were incubated at 37°C with 5% CO2, in a humidified incubator and passaged when confluence reached 80%.
Proliferation assay. CCA cells were seeded into 96-well plates at 3×103 cells/well and cultured overnight in a humidified incubator at 37°C with 5% CO2. Cells were then treated with 5, 10, 20, 40 mM metformin for 48 h or for different times. The number of viable cells was determined using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay as described in our previous study (14).
Migration and invasion assay. CCA cells were pretreated with 10 mM metformin for 24 h prior to migration and invasion assays according to our previous report (14). Cells were allowed to migrate in the absence of metformin for 12 h for KKU-M213 and 24 h for KKU-M214 cells. The invasion assay was performed using the same protocol as the migration assay except that the upper chamber was pre-coated with 50 μg/well of Matrigel (BD Bioscience, Bedford, MA, USA).
Anoikis assay. CCA cells were suspended in a complete medium containing 0, 10, 20 mM of metformin and then seeded into a 24-well ultralow attachment plate (Corning, Lowell, MA, USA) which was coated with a neutral charge of hydrogel to prevent the adherence of cells. Cells (5×104 cells/well) were then incubated in the non-adhered condition for 48 h. Total cells in each well were collected, washed twice with PBS and treated with trypsin for 5 min to obtain a single-cell suspension. The numbers of viable and dead cells were determined using the trypan blue dye exclusion assay. Three independent experiments were performed with triplicated tests.
Western blot analysis for cell-cycle proteins. CCA cells were treated with 10, 20, 40 mM metformin for 48 h whereas those for protein markers of metastasis were treated with 10 mM metformin for 24 h. Cells were lysed with NP-40 lysis buffer containing protease and phosphatase inhibitor cocktail (Roche, Mannheim, Germany). Cell lysates were prepared, subjected to sodium dodecyl sulphate -polyacrylamide gel electrophoresis and transferred to a Hybond™-polyvinylidene fluoride (PVDF) membrane as mentioned previously (14). The membrane was probed with each primary antibody at 4°C overnight and with horseradish peroxidase-conjugated secondary antibody for 1 h at room temperature. Signals were detected using an enhanced chemiluminescence prime western blotting detection kit (GE Healthcare) and the image was analyzed using Image Quant™ Imager (GE Healthcare).
Immunocytofluorescence analysis. CCA cells (8×103 cells/well) were cultured in Matrigel pre-coated slide chambers overnight at 37°C with 5% CO2. The cells were treated 10 and 20 mM metformin for 24 h. Immunofluorescent staining was performed and recorded as described previously (14). Fixed cells were incubated with 1:100 anti-STAT3 or anti-p65 antibodies at 4°C overnight. Then, 1:200 FITC-conjugated secondary antibody for STAT3 or 1:200 Alexa Fluor 568-conjugated secondary antibody for p65 antibody and 1:10,000 of Hoechst 33342 were applied at room temperature for further 2 h. The immunofluorescent images were captured and analyzed using Nikon NIS-Elements software (Nikon, Tokyo, Japan).
Statistical analysis. All quantitative data are reported as mean±SD. Triplicate tests for each experiment and at least two independent experiments were performed. The differences between two groups were compared using the two-tailed Student's t-test. A value of p<0.05 was considered as statistically significant.

Metformin exerted an antiproliferative effect on cholangiocarcinoma (CCA) cell lines in a dose- and time-dependent manner. Cells were treated with different concentrations of metformin for different times and the viable cells were determined using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. A: Cells were cultured in the presence of 5, 10, 20 or 40 mM metformin for 48 h. B: CCA cells were treated with 10 or 20 mM metformin for 24, 48 and 72 h. Data are mean±SD of three independent experiments. *Significantly different from control of each time point, p<0.05. C: Metformin had similar growth inhibitory effects on CCA cells regardless of the glucose level in the medium. Data are the mean±SD from triplicate assays from one of two independent experiments (*p<0.05). NG: Normal glucose; HG: high glucose; Met: metformin.
Results
Metformin suppressed proliferation of CCA cells regardless of glucose level. The effect of metformin on cell proliferation was first tested in four CCA cell lines cultured in medium with 10-40 mM of metformin for 48 h. The antiproliferative effect of metformin was observed in all CCA cell lines tested in a dose- (Figure 1A) and time- (Figure 1B) dependent manner. All CCA cell lines responded to metformin treatment in a similar manner, with a half-maximal inhibitory concentration (IC50) that ranged from 14.3 to 18.6 mM (Table I).
The half-maximal inhibitory concentration (IC50) of metformin for cholangiocarcinoma cell lines after 48 h treatment.
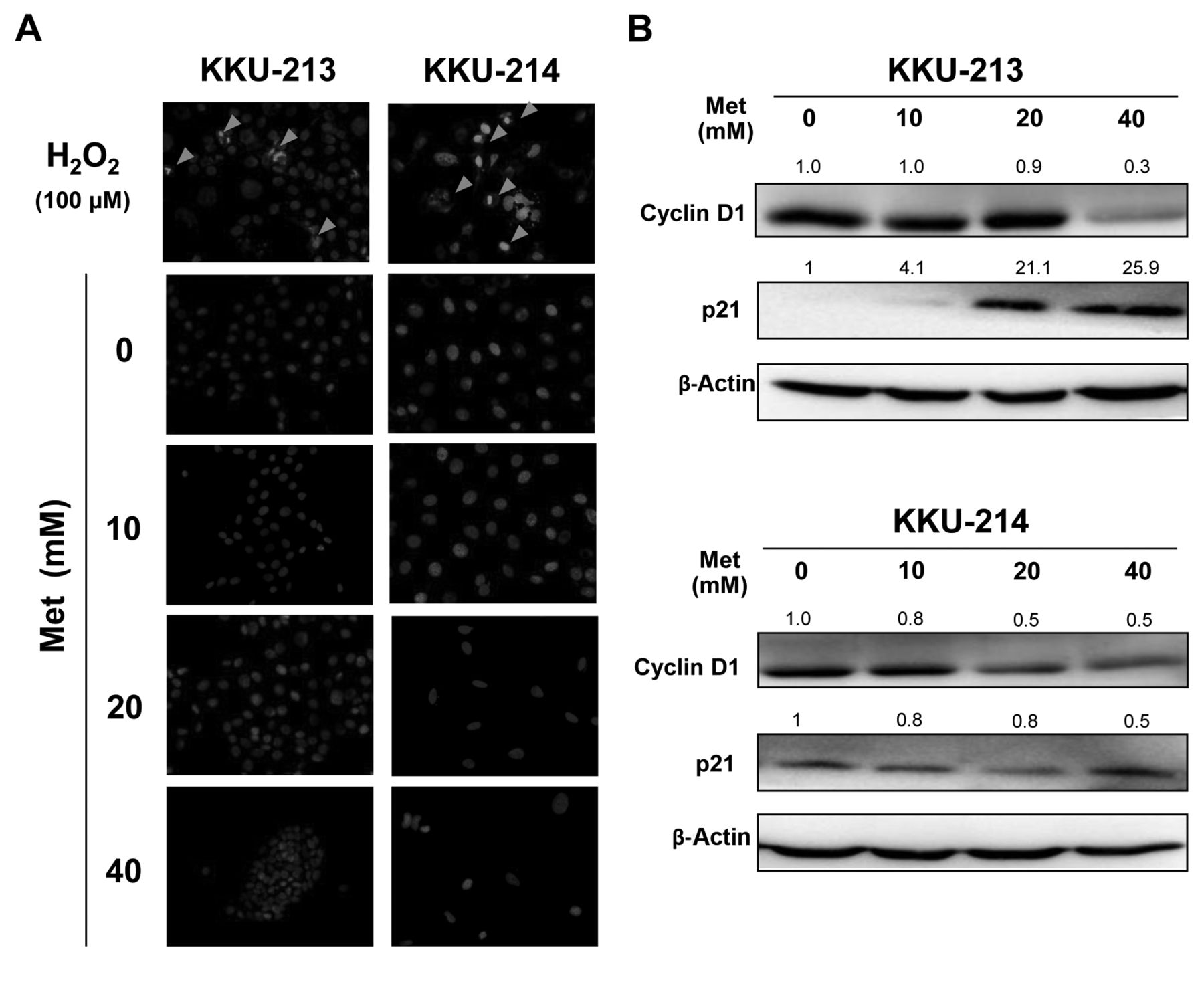
Metformin inhibited cholangiocarcinoma cell growth without induction of apoptosis. A: Hoechst staining was used to demonstrate nuclear morphology and chromatin condensation of DNA. Cells were treated with 10-40 mM of metformin for 48 h. No chromatin condensation was noted in control nor in metformin-treated cells. H2O2 (100 μM) was used as a positive control to illustrate the feature of apoptotic nuclei (indicated by arrows). B: Western blot analysis of cell-cycle regulatory proteins: cyclin D1 and p21. Quantification of each protein was performed using β-actin as an internal control and compared by designating expression of the untreated sample as 1. Data are representative of two independent experiments. Met: Metformin.
As DM and CCA seem to be associated, and metformin is generally used for diabetic patients who have chronic hyperglycemia, it was next tested as to whether the antiproliferative effect of metformin would be effective in patients with normal blood glucose. The antiproliferative effect of metformin was then tested in KKU-055 and KKU-213 cells cultured in media with normal glucose or high glucose, in the presence or absence of metformin. The results showed that regardless of glucose concentration, metformin exhibited similar effects on growth inhibition in a dose-dependent manner (Figure 1C).
As apoptotic cells were not observed in metformin-treated cells, it was hypothesized that the antiproliferative activity of metformin should be, partly, due to growth suppression rather than apoptosis induction. To demonstrate this, KKU-213 and KKU-214 cells were treated with different concentrations of metformin for 48 h and apoptotic cells were observed using Hoechst staining. Cells treated with 100 μM hydrogen peroxide for 48 h were used as positive controls. As shown in Figure 2A, nuclear fragmentation and chromatin condensation were observed in the hydrogen peroxide-treated cells but were rarely observed in the cells treated with up to 40 mM metformin.
To affirm the cytostatic effect of metformin, cell-cycle regulatory proteins, namely cylclin D1 and p21, were determined in cells treated with metformin for 48 h. Compared to the untreated control cells, suppression of cyclin D1 and up-regulation of p21 were observed in CCA cells treated with increasing metformin concentrations (Figure 2B).

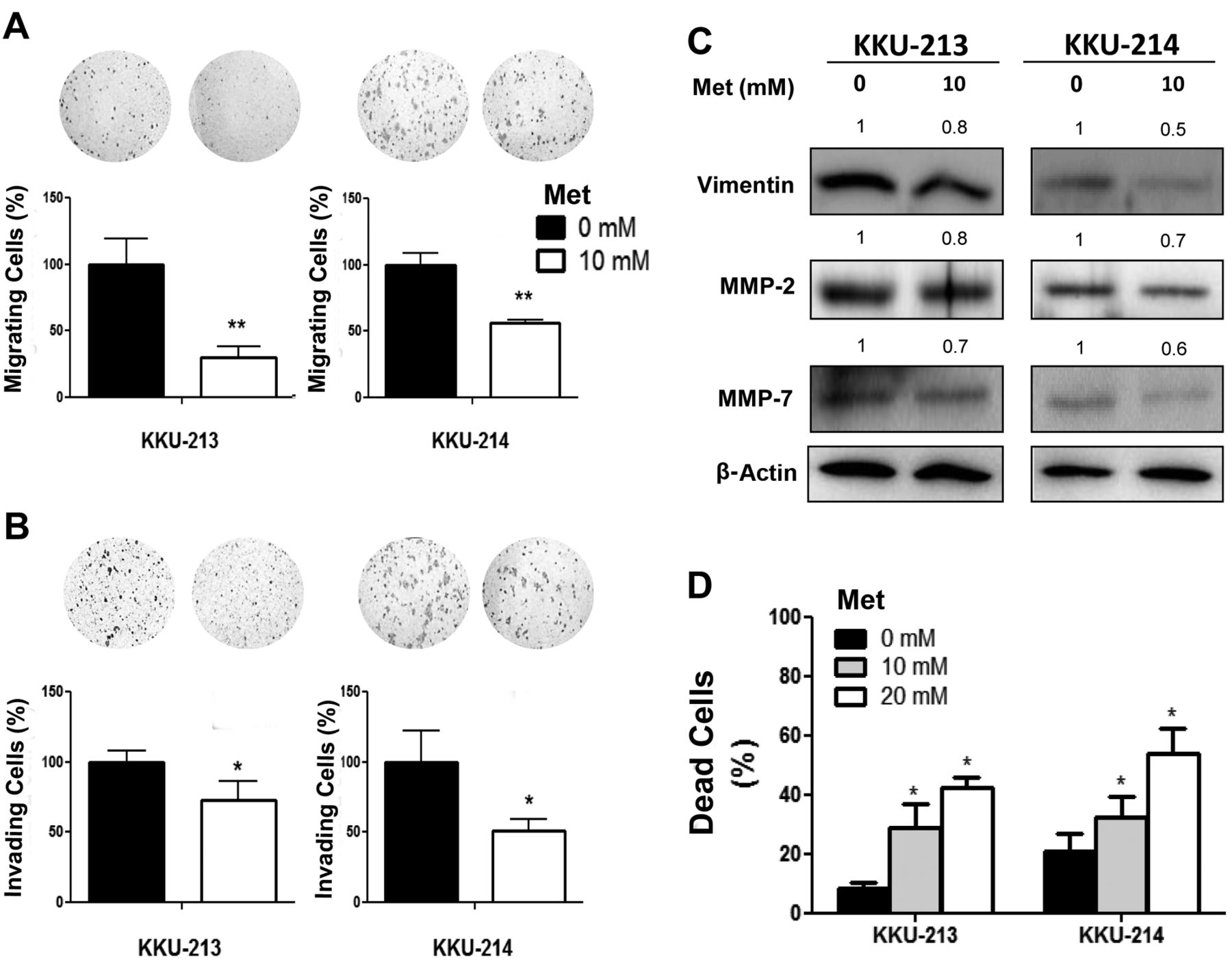
Metformin suppressed migration and invasion and induced anoikis of cholangiocarcinoma cells. KKU-213 and KKU-214 cells were pretreated with 10 mM metformin for 24 h before being subjected to migration and invasion assays. Numbers of migrating (A) and invading (B) cells were counted and compared by assigning the controls a value of 100%. Data are the mean±SD of triplicates from one of two independent experiments. C: Western blots of vimentin, matrix metalloproteinase (MMP)-2 and -7. Quantification of each protein was performed using β-actin as an internal control and compared by designating expression of the untreated sample as 1. D: Anoikis resistance was assessed in the absence and presence of 10 or 20 mM metformin for 48 h. The number of dead cells was counted using the trypan blue dye exclusion assay. The data are representative of three independent experiments. *p<0.05, **p<0.01.
Low dose of metformin inhibited migration and invasion of CCA cells. CCA cells (KKU-213 and KKU-214) treated with 10 mM metformin for 24 h were used to examine the effect of metformin on migration and invasion, as this concentration did not significantly affect growth of CCA cells. After 24 h of metformin treatment, cells were allowed to migrate in the absence of metformin for 12 h for KKU-M213 and 24 h for KKU-M214. Metformin significantly reduced migrated cells to 30-50% and invaded cells to 50-70% of the controls (Figure 3A and B). To explore the underlying mechanism, the expression levels of vimentin, MMP2 and MMP7 were determined using western blotting. As shown in Figure 3C, the expressions of vimentin, MMP2 and MMP7 were reduced in metformin-treated compared to control cells.
Metformin induced anoikis of CCA cells. Anoikis resistance or resistance to apoptosis in non-adherent conditions is a key property of cells that metastasize. Whether metformin can induce anoikis was next analyzed in CCA cells treated with 10 and 20 mM metformin in a 24-well ultralow attachment plate for 48 h. The total cells were harvested and the live vs. dead cells were determined using the trypan blue dye exclusion assay. As shown in Figure 3D, the basal percentage of dead cells induced by anoikis in the control cells was 10-20% of the total cell count. Metformin at 10 and 20 mM, however, significantly induced cell death by anoikis by up to 2- to 3-fold of that observed in the control cells.
Metformin activated AMPK. It is well documented that the conventional effect of metformin is through AMPK activation. The level of phosphorylation AMPKα, a stimulatory subunit of AMPK, were determined in CCA cells treated with 10 and 20 mM metformin for 48 h. The western blot results demonstrated that p-AMPKα was increased 3- to 4-fold in metformin-treated cells while total AMPKα remained unaltered (Figure 4A). The ratio of p-AMPKα to AMPKα of both cell lines increased respectively in metformin-treated cells.
Metformin inhibited nuclear localization of NF-ĸB and STAT3 without affecting their activations. Since the proteins affected by metformin treatment, as observed in this study, can be mediated by NF-ĸB and STAT3, important transcription factors in carcinogenesis and progression of CCA (15, 16), it was thus interesting to investigate whether metformin affected CCA cells via NF-ĸB or STAT3 pathways. The levels of NF-ĸB (p65) and STAT3 from whole-cell lysates of CCA cells treated with 10 and 20 mM metformin for 48 h and the control cells were determined using western blots. Under metformin treatment, the expression of NF-ĸB (p65) slightly increased in KKU-213 cells, but was unaltered in KKU-214 cells (Figure 4B). The western blots of STAT3 indicated that activation of STAT3 determined as the ratio of p-STAT3 (Y705 and S727) to STAT3 of the metformin-treated cells did not differ from that of the control cells (Figure 4B).
As nuclear translocation is the crucial step for NF-ĸB and STAT3 activation, the localization of NF-ĸB (p65) and STAT3 was investigated using immunocytofluorescent staining and nuclei were localized using Hoechst staining. These generated bright blue signals for NF-ĸB p65 and bright purple signals for STAT3 when these were localized in the nucleus. As demonstrated in Figure 4C, the signals of NF-ĸB (p65) and STAT3 of almost all of the control cells were localized in the nucleus. In contrast, the signals of NF-ĸB (p65) and STAT3 of the metformin-treated cells were mainly in the cytoplasm. These results indicate an inhibitory effect of metformin on the nuclear translocation of NF-ĸB (p65) and STAT3 in these CCA cell lines.
Discussion
Metastasis is a leading cause of death in all patients with cancer. It involves many biological processes such as invasion and migration to adjacent tissues and blood vessels, survival in the blood circulation and adhesion to secondary sites. Accumulated evidence suggests metformin to be a potential drug for suppressing the progression of tumors in many cancer types (5). The present study demonstrates the effects of metformin on the inhibitory mechanisms of Opisthorchis viverrini-associated CCA cells. Metformin exhibited dose- and time-dependent antiproliferative effects on CCA cells. In this study, it was additionally demonstrated for the first time that metformin at a non-antiproliferative dose significantly suppressed cell migration and invasion and the underlining mechanism was shown to be via the inhibition of nuclear translocation of NF-ĸB and STAT3.
In the current study, metformin effectively suppressed proliferation of CCA cell lines in a time- and dose-dependent manner. Metformin affected proliferation of CCA cells regardless of their chemo- or radioresistant behavior. The IC50 of all four CCA cell lines tested were similar in the range of 14.3-18.6 mM. KKU-100, a chemoresistant cell line (17) and KKU-055, a radioresistant clone (18), responded to metformin in the same manner. These effective doses were in agreement with reports on non-liver fluke-associated CCA (12, 19). Metformin also affected proliferation of CCA cells irrespectively of the glucose condition. DM has been considered as a common risk factor for various cancer types, including CCA (7). In addition, a high glucose condition can significantly enhance proliferation, migration and invasion of CCA cells (14). In the present study, metformin reduced proliferation not only of CCA cells cultured in normal glucose medium but also those cultured in high glucose medium. This information may imply an antitumor effect of metformin in patients with CCA regardless of diabetic condition.
Metformin inhibited proliferation of CCA cells without induction of apoptosis, as no obvious apoptotic cells were observed with metformin treatment. In addition, the western blot analyses indicated an increase of p21 and suppression of cyclin D1 in metformin-treated cells. These observations were in agreement with those previously reported in many cancer cell types (13, 20), including non-liver fluke-associated CCA cells (12, 19). In addition to cell-cycle regulatory proteins, the alteration of microRNA expression in metformin treated CCA cells was also noted (12, 21).
The present study also demonstrated, as far as we are aware of for the first time, the inhibitory effect of metformin on the metastatic potential of CCA cells. At low doses, metformin significantly suppressed migration and invasion, and induced anoikis of CCA cells. The molecular basis underlying these observations in metformin-treated cells was shown to be via suppression of vimentin, a marker of epithelial–mesenchymal transition related to cell motility, and anoikis, along with MMP2 and MMP7, essential enzymes for extracellular matrix degradation. These three markers are known to be under regulation of several key pathways, including STAT3 and NF-ĸB (22).

Metformin increased phosphorylated-5’ adenosine monophosphate activated protein kinase (p-AMPK) and inhibited nuclear localization of nuclear factor-kappa B (NF-ĸB) and signal transducer and activator of transcription 3 (STAT3). Cholangiocarcinoma cells were cultured in the absence or presence of 10 or 20 mM metformin for 48 h before western blot analysis. A: p-AMPKα and AMPKα, B: total NF-ĸB (p65), and C: p-STAT3 (Y705 and S727) and STAT3. Quantification of each protein was performed using β-actin as an internal control and compared by designating expression of the untreated sample as 1. D: Cells were treated with 10 or 20 mM metformin for 24 h and the cellular localizations of NF-ĸB (p65) (green) and STAT3 (red) were observed using immunocytofluorescent staining, ×400 magnification. Met: Metformin.
The conventional effect of metformin is trough the indirect activation of AMPK, however, there are many consequent transcription factors and signaling pathways activated/inactivated in responses to AMPK activation (5). Apart from AMPK, metformin exerts anticancer effects via multiple pathways, e.g. inhibition of the mTOR pathway in CCA (19), a decrease of forkhead box protein M1 (FOXM1) in leukemia (23), suppression of STAT3 activation in lung adenocarcinoma (24)and in triple-negative breast cancer (25), and inactivation of STAT3 and NF-ĸB in several cancer cell lines (26, 27).
In the present study, metformin activated the AMPK pathway as the level of p-AMPK in CCA cells actually increased in metformin-treated cells. To investigate the alternative pathways in response to metformin treatment, NF-ĸB and STAT3, the major transcription factors reported in development and progression of liver fluke-associated CCA (15, 16) were further examined. The results from western blots of whole-cell lysates showed no differences of p-STAT3 at both Y705 and S727 and the expression levels of p65 between the controls and the metformin-treated cells. This indicates that metformin had no effect on STAT3 phosphorylation and NF-ĸB expression. In contrast, the strong reduction of nuclear localization of STAT3 and p65 was observed in metformin-treated cells. Nuclear translocation of STAT3 and p65 is an important step to complete the activation of both transcriptional factors. Suppression of nuclear translocation of STAT3 and p65 may reduce the expression of their downstream regulated genes, e.g. vimentin, MMP2 and MMP7, as shown in this study. These assumptions are supported by several previous reports showing that inhibition of STAT3 and NF-ĸB activation using specific inhibitors significantly suppressed growth (14, 16), migration and invasion (28) of CCA cells.
The effects of metformin on the activation of STAT3 and the expression of NF-ĸB have been reported in many studies (24, 25, 29-31). In the present study, these effects of metformin were not observed, however, the effects of metformin on nuclear translocation of STAT3 and NF-ĸB were pronounced. This effect of metformin on nuclear translocation of NF-ĸB (p65) was reported for the first time in the present study.
Taken together, the findings of this study indicate the additional effect of metformin on anti-metastatic properties of CCA cell lines. This action of metformin concurs with the observation of longer survival of patients with CCA with DM who received metformin than those without metformin treatment (21). The findings from this study are important in repurposing metformin as a drug of choice for CCA treatment, as metformin has several advantages in the treatment of CCA: i) Metformin affects CCA cells regardless of their etiology. Metformin effectively suppressed growth of liver fluke-associated and non-liver fluke-associated CCA. ii) Metformin effectively inhibited growth of CCA cells unrelated to the background of chemo- or radioresistant behavior: both a chemoresistant CCA cell line, KKU-100 and a radioresistant clone, KKU-055, were sensitive to metformin treatment in a similar manner. iii) A low dose of metformin can suppress metastatic activity of CCA cells. iv) Lastly, because metformin is already used in clinical practice, its safety and efficacy are known. Since CCA is a highly metastatic and highly recurrent cancer, the antimetastatic activity of metformin may be of an advantage for patients with CCA with metastasis. Further studies in animal models should be conducted to ensure the efficacy of the anticancer effect of metformin, either as a single drug or as a part of a combined treatment for CCA.
Acknowledgements
This work was co-supported by research grants from Khon Kaen University and the TRF Senior Research Scholar Grant, Thailand Research Fund to S. Wongkham (RTA5780012), a Khon Kaen University Research grant (KKU59) to U. Cha'on and a research grant from the Junior Science Talent Project (JSTP), National Science and Technology Development Agency (NSTDA) to C. Saengboonmee (JSTP-06-56-01E). C. Seangboonmee also thanks Faculty of Medicine, Khon Kaen University for the support of the BSc-PhD-MD program. We would like to thank Professor James A. Will for editing this article via the Faculty of Medicine Publication Clinic, Khon Kaen University, Thailand.
Footnotes
This article is freely accessible online.
- Received November 4, 2016.
- Revision received November 25, 2016.
- Accepted November 29, 2016.
- Copyright© 2017 International Institute of Anticancer Research (Dr. John G. Delinassios), All rights reserved
References
In this issue





{kind=link}
{kind=link}
{kind=link}
{kind=link}
Jump to section
Related Articles
Cited By...
- Repurposing Metformin for Cancer Treatment: A Great Challenge of a Promising Drug
- Artemisinin mimics nitric oxide to reduce adipose weight by targeting mitochondrial complexes
- An Improved In Vivo Methodology to Visualise Tumour Induced Changes in Vasculature Using the Chick Chorionic Allantoic Membrane Assay
- Metformin-associated Chemopreventive Effects on Recurrence After Hepatic Resection of Hepatocellular Carcinoma: From In Vitro to a Clinical Study
- Anti-proliferative, Cytotoxic and NF-ĸB Inhibitory Properties of Spiro(Lactone-Cyclohexanone) Compounds in Human Leukemia